INTRODUCTION
Le syndrome combiné polypose juvénile/télangiectasie hémorragique héréditaire (combined syndrome of juvenile polyposis and hereditary haemorrhagic telangiectasia, JP–HHT Syndrome) est décrit pour la première fois en 2004 par Gallione et al. (1). Les individus atteints présentent les manifestations cliniques des deux pathologies et sont exposés à un risque augmenté de cancer gastro-intestinal.
Cette entité rare est causée par des mutations dans le gène MADH4, qui code pour une protéine, SMAD4, impliquée dans la voie de signalisation TGF-β (1). La prévalence de ce syndrome n’est pas connue. Toutefois, approximativement 20% des individus atteints de la polypose juvénile sont mutés pour ce gène (2). Et seuls 1 à 2% des patients avec la maladie d’Osler-Rendu-Weber sont mutés pour MADH4 (3).
Le but de cet article est de présenter ce syndrome peu connu et de souligner les implications qui en découlent dans la prise en charge des patients atteints de la polypose juvénile ou de la maladie d’Osler-Rendu-Weber.
CAS CLINIQUE
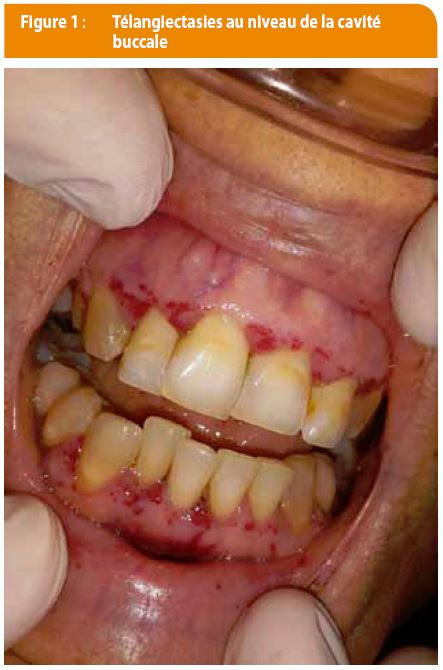
Une patiente âgée de 41 ans, et atteinte de la HHT, est suivie en gastro-entérologie pour anémie ferriprive chronique. Depuis l’âge de 16 ans, elle présente des télangiectasies au niveau de la langue, des lèvres et des gencives (Figures 1 et 2) ainsi que des épistaxis quotidiennes et des gingivorragies. La patiente a subi plusieurs cautérisations électriques pour ses épistaxis, une pilule à base d’éthinylestradiol et de lévonorgestrel (Microgynon 50 ®) a également été instaurée. Malgré ces mesures, les saignements ne sont pas contrôlés et sont responsables d’une anémie ferriprive chronique qui justifie des injections intraveineuses de fer mensuellement. Le taux d’hémoglobine est maintenu entre 9 et 10g/dl.

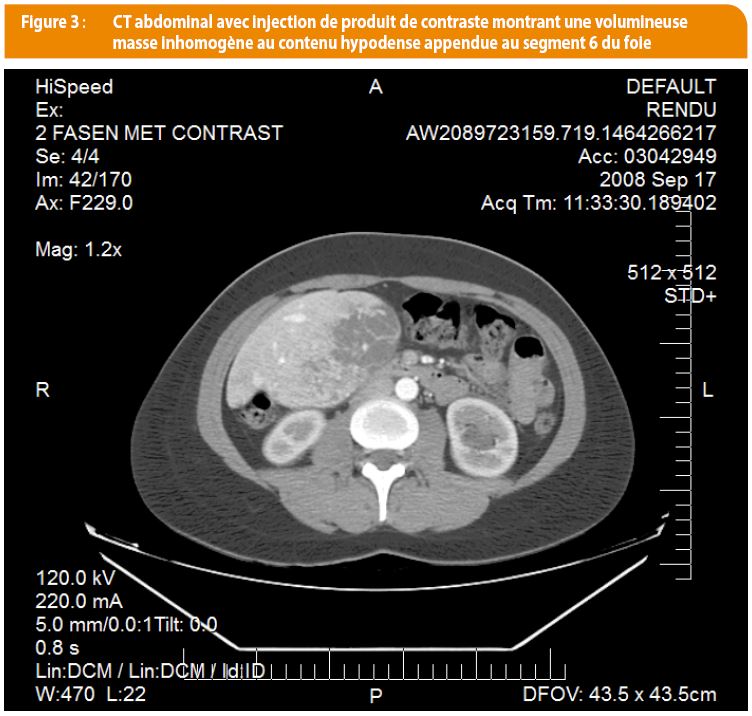
En 2008, la patiente se présente à la consultation pour une douleur brutale à l’hypochondre droit. Cliniquement, on note une masse palpable au niveau du lobe droit du foie. La biologie montre une anémie ferriprive : Hb 9,8 g/dl, Ferritine 4 µg/l (normale : 10-270µg/l) Volume globulaire moyen 75 µ3 (normale : 85-95µ3) avec des GGT augmentées à 177 U/L (normale : 11-50 U/L). Face à ce tableau, une IRM hépatique et un scanner abdominal sont réalisés. Ceux-ci objectivent une masse appendue au lobe droit du foie avec un contenu inhomogène en son centre (Figure 3). En outre, on visualise des vaisseaux hépatiques tortueux et des malformations artério-veineuses, témoins de la HHT. Un adénome compliqué, une thrombose d’une malformation artério-veineuse ou encore une pathologie tumorale sont suspectés. Une résection laparoscopique a lieu et l’analyse histologique confirme un adénome hépatocellulaire. En raison de cette découverte, la pilule oestro-progestative est arrêtée.
En 2010, une exploration digestive est réalisée. La gastro-duodénoscopie montre des lésions angiodysplasiques au niveau de l’estomac et du duodénum. Celles-ci sont traitées par coagulation au plasma d’argon (APC). La colonoscopie est sans particularité. Aucune hémorragie n’est visualisée.
Un an plus tard, la patiente est référée au Centre de Référence pour la maladie d’Osler-Rendu à Lyon où une analyse génétique à la recherche d’une mutation dans le gène MADH4 est effectuée. L’analyse révèle que la patiente est porteuse d’une mutation dans ce gène. Les experts recommandent une surveillance régulière du tractus digestif. L’examen endoscopique de l’estomac, de l’intestin grêle et du côlon réalisé cette année-là s’avère négatif.
En 2014, on procède à un nouveau dépistage. La gastro-duodénoscopie montre plusieurs polypes d’aspect bénin localisés dans le duodénum. Le plus gros est biopsié et se révèle être un polype hyperplasique. La colonoscopie quant à elle, met en évidence deux polypes au niveau du côlon transverse qui sont enlevés ainsi qu’une troisième lésion polypoïde de plusieurs centimètres, non résécable, à hauteur de la valvule iléo-caecale. L’analyse anatomopathologique montre que les polypes coliques sont des hamartomes, la biopsie du caecum met en évidence un adénocarcinome.
Le bilan d’extension confirme la présence d’un processus tumoral au niveau du caecum, sans métastase à distance. Le dosage du CEA est normal.
Une hémicolectomie droite par coelioscopie est alors décidée. L'examen histologique de la pièce opératoire décèle un adénocarcinome intra-muqueux de 3,5 cm de diamètre. Les marges de résection sont saines. 25 ganglions sont réséqués, aucun d’entre eux n’est envahi. Il n’y a pas de perméation lymphatique. Le stade tumoral établi est pTisN0M0.
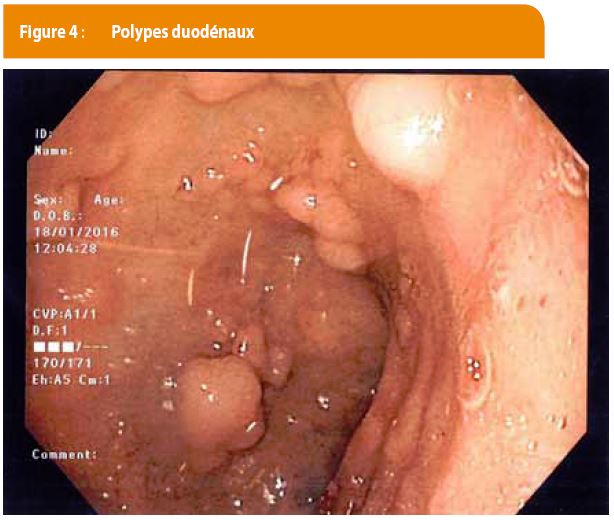
En janvier 2016, un premier contrôle post-opératoire ne montre pas de récidive de polypes à la colonoscopie. La gastro-duodénoscopie révèle plusieurs polypes duodénaux sans malignité (figure 4). Un second contrôle, un an plus tard, se révèle rassurant.
DISCUSSION ET CONCLUSION
La HHT est une dysplasie vasculaire à transmission autosomique dominante qui affecte une personne sur 5000 à 8000 (4-5). Elle se caractérise par des épistaxis, des télangiectasies cutanéo-muqueuses et des malformations artério-veineuses (MAV) viscérales. Son expressivité est variable et sa pénétrance est liée à l’âge (6).
L’épistaxis est la manifestation la plus fréquente et atteint la grande majorité des patients. Elle est spontanée, récidivante et est due aux télangiectasies présentes dans la muqueuse nasale. Ces saignements peuvent mener, comme dans le cas présenté, à une anémie chronique nécessitant une supplémentation en fer ou des transfusions (7).
Les télangiectasies sont multiples et typiquement localisées sur la face, les lèvres, la cavité buccale, le nez et les doigts (6). Elles peuvent également être localisées tout au long du tractus gastro- intestinal où elles peuvent causer des hémorragies se manifestant en général à partir de 50-60 ans (8). L’atteinte gastro-intestinale se manifeste moins fréquemment par des angiodysplasies, ce qui fut le cas pour notre patiente, ou des MAV (7).
Les malformations artério-veineuses sont présentes principalement dans le poumon, le foie, le système nerveux central et le tractus gastro-intestinal (3).
Le diagnostic de HHT est un diagnostic clinique basé sur les critères de consensus international de Curaçao (9).
- Épistaxis récidivantes et spontanées.
- Télangiectasies multiples localisées à des endroits typiques (lèvres, cavité orale, doigts, nez).
- Lésions viscérales (télangiectasies gastro-intestinales avec ou sans saignements, mav pulmonaire, hépatique, cérébral, spinal).
- Histoire familiale positive d’HHT (un apparenté du premier degré avec HHT).
Le diagnostic est certain lorsqu’au moins trois critères sont remplis. Il est possible ou suspecté en présence de deux critères et improbable lorsque moins de deux critères sont présents.
Le dépistage des MAV pulmonaires et cérébrales est recommandé chez tous les patients avec un diagnostic possible ou confirmé d’HHT étant donné les complications mortelles qu’elles peuvent engendrer (10).
Notre patiente présente une HHT avec épistaxis, télangiectasies cutanéo-muqueuses et atteintes viscérales. Notons que l’anamnèse familiale est négative pour la HHT.
L’atteinte hépatique reste asymptomatique chez la grande majorité des patients (11) et se caractérise par des shunts intra hépatiques (12) : trois types de shunts peuvent être observés: shunt artério-veineux (entre l’artère hépatique et les veines hépatiques) shunt artério-portal (entre l’artère hépatique et la veine porte) et shunt porto-veineux (entre le tronc porte et les veines hépatiques). Ces shunts peuvent se manifester par une insuffisance cardiaque à haut débit, une hypertension portale, une cholangiopathie ischémique ou encore une encéphalopathie. L’atteinte hépatique peut également causer une hyperplasie nodulaire régénérative ou hyperplasie nodulaire focale (12).
L’adénome hépatocellulaire n’a pas été associé à la HHT dans la littérature. Nous pensons donc que cet adénome a été induit par la pilule oestro-progestative (Microgynon 50®) que notre patiente a prise pendant une quinzaine d’années. En effet, l’adénome hépatocellulaire est fortement lié à la prise de contraceptifs oraux et le risque de développer un adénome hépatocellulaire dépend de la dose et la durée d’utilisation (13). L’incidence de l’adénome hépatocellulaire chez les femmes prenant une contraception orale hautement dosé en œstrogène a été estimée dans les années 70 à 3,4 pour 100 000 (13). Toutefois, ce chiffre est sans doute diminué depuis l’introduction de pilules plus faiblement dosées en œstrogène (14).
Jameson et Cave (15) recommandent l’emploi d’un traitement oral oestro-progestatif aux doses contraceptives pour les épistaxis chez la femme en âge de fertilité. Certes, un essai contrôlé randomisé en double aveugle (16) a montré une diminution significative des hémorragies gastro-intestinales sévères chez les patients traités par oestro-progestatif. Cependant, concernant les épistaxis, seul un essai clinique non contrôlé (17) a montré une amélioration après traitement. Remarquons que l’hormonothérapie n’a pas été efficace pour notre patiente.
Trois gènes responsables ont été identifiés dans la HHT : ENG (18), ACVRL1 (19), MADH4 (1). Ces gènes codent tous pour des protéines impliquées dans la voie de signalisation TGF-β. Les mutations dans le gène MADH4, qui code pour le facteur de transcription SMAD4, sont retrouvées chez 1 à 2% des individus avec HHT. Les mutations de novo sont plus fréquentes (3).
La polypose juvénile (Juvenile polyposis, JP) est aussi une maladie autosomique dominante qui touche une personne sur 100 0000 (20). Elle se présente habituellement durant l’enfance et est caractérisée par des polypes hamartomateux dans le tractus gastro-intestinal. Ces polypes sont principalement localisés dans le côlon. Signalons qu’on peut retrouver des polypes hyperplasiques dans l’intestin grêle (21-22).
Leur nombre varie entre cinq et plusieurs centaines (23). La polypose juvénile se manifeste par des rectorragies, de l’anémie, des douleurs abdominales, de la diarrhée, une invagination ou encore une obstruction intestinale ou un prolapsus (24). Deux gènes causaux ont été identifiés : MADH4 (25) et BMPR1A (26).
Le diagnostic de la polypose juvénile est établi si l’un des critères suivants est rempli (27) :
- au moins 5 polypes juvéniles au niveau colorectal ;
- multiples polypes juvéniles répartis à travers le tractus gastro-intestinal ;
- polype(s) juvénile(s) chez un individu avec une histoire familiale de polypose juvénile.
La polypose juvénile est associée à un risque accru de cancer gastro-intestinal (28) et une surveillance par colonoscopie et endoscopie haute est recommandée (29).
Plusieurs cas ont rapporté l’association de la HHT et de la polypose juvénile chez un même patient (30-32) mais ce n’est qu’en 2004 que Gallione et al. (1) ont démontré que certaines mutations de SMAD4 produisent un syndrome combinant polypose juvénile et télangiectasie hémorragique héréditaire. Plus tard, Gallione et al. (32) rapportèrent que n’importe quelle mutation dans le gène MADH4 pouvait causer ce syndrome. Les patients avec JP/HHT ont les symptômes des deux maladies mais des critères diagnostiques précis n’ont pas été définis. Cette découverte a motivé les experts de Lyon à rechercher une mutation dans ce gène chez notre patiente, ce qui a permis de détecter précocement un adénocarcinome du côlon.
La génétique a une implication directe pour la prise en charge des patients atteints de la JP ou de la HHT (33). En effet, les patients HHT avec la mutation SMAD4 courent un risque de syndrome JP-HHT et de cancer colorectal. Ils nécessitent une surveillance endoscopique. De même, seuls les patients atteints de la polypose juvénile et mutés pour SMAD4 doivent être dépistés pour les MAV viscérales associées à HHT. Gallione et al. (33) recommandent donc de procéder à une analyse génétique pour chaque patient atteint de la polypose juvénile ou de la télangiectasie hémorragique héréditaire.
Signalons que récemment, plusieurs cas ont rapporté la présence de pathologies aortiques chez des patients JP-HHT (34-35).
En conclusion, ce cas clinique illustre un syndrome rare et souligne l’importance de référer les patients atteints d’une maladie rare à un centre de référence pour une prise en charge optimalisée.
RECOMMANDATIONS PRATIQUES
En cas de pathologie rare, nous recommandons de référer le patient à un centre de référence pour sa maladie.
Chez tout patient atteint de la polypose juvénile ou de la maladie d’Osler-Rendu-Weber, il est important de disposer d’une analyse génétique afin d’identifier ceux qui sont susceptibles de développer le syndrome JP-HHT.
AFFILIATIONS
1. Étudiante en Master 4, Université catholique de Louvain.
2. Chef de service, service de gastro-entérologie, AZ Groeninge, 8500 Kortrijk.
CORRESPONDANCE
Edeline Kaze edeline.kaze@student.uclouvain.be
RÉFÉRENCES
(1) Gallione CJ, Repetto GM, Legius E, Rustgi AK, Schelley SL, Tejpar S,et al. A combined syndrome of juvenile polyposis and hereditary haemorrhagic telangiectasia associated with mutations in MADH4 (SMAD4). Lancet 2004; 363 (9412): 852–859.
(2) Howe JR, Sayed MG, Ahmed AF, Ringold J, Larsen-Haidle J,Merg A, et al. The prevalence of MADH4 and BMPR1A mutations in juvenile polyposis and absence of BMPR2, BMPR1B, and ACVR1 mutations. J Med Genet 2004; 41 (7):484–491.
(3) Gallione CJ, Richards JA, Letteboer TG, Rushlow D, Prigoda NL, Leedom TP, et al. SMAD4 mutations found in unselected HHT patients. J Med Genet 2006; 43 (10):793–797.
(4) Dakeishi M, Shioya T, Wada Y, Shindo T, Otaka K, Manabe M, et al.Genetic epidemiology of hereditary haemorrhagic telangiectasia in a local community in the northern part of Japan. Hum Mutat 2002; 19 (2): 140–148.
(5) Kjeldsen AD, Vase P, Green A. Hereditary haemorrhagic telangiectasia: a population-based study of prevalence and mortality in Danish patients. J Intern Med 1999; 245 (1): 31–39.
(6) Plauchu H, de Chadarévian JP, Bideau A, Robert JM. Age-related profile of hereditary hemorrhagic telangiectasia in an epidemiologically recruited population. Am J Med Genet 1989; 32 (3):291–297.
(7) Guttmacher AE, Marchuk DA, White RI., Jr Hereditary hemorrhagic telangiectasia. N Engl J Med 1995; 333 (14):918–924.
(8) Begbie ME, Wallace GMF, Shovlin CL. Hereditary haemorrhagic telangiectasia (Osler-Weber-Rendu syndrome): a view from the 21st century. Postgrad Med J 2003; 79 (927):18–24.
(9) Shovlin CL, Guttmacher AE, Buscarini E, Faughnan ME, Hyland RH, Westermann CJ et al. Diagnostic criteria for hereditary hemorrhagic telangiectasia (Rendu-Osler-Weber syndrome). Am J Med Genet 2000; 91 (1): 66–67.
(10) Faughnan ME, Palda VA, Garcia-Tsao G, Geisthoff UW, McDonald J, Proctor DD, et al. International guidelines for the diagnosis and management of hereditary haemorrhagic telangiectasia. J Med Genet 2011 ; 48 (2):73–87.
(11) Ianora AA, Memeo M, Sabba C, Cirulli A, Rotondo A, Angelelli G. Hereditary hemorrhagic telangiectasia: multi-detector row helical CT assessment of hepatic involvement. Radiology 2004; 230 (1):250–259.
(12) G. Garcia-Tsao, Liver involvement in hereditary hemorrhagic telangiectasia (HHT). J Hepatol 2007; 46 (3): 499–507.
(13) Rooks JB, Ory HW, Ishak KG, Strauss LT, Greenspan JR, Hill AP et al . Epidemiology of hepatocellular adenoma. The role of oral contraceptive use. JAMA 1979; 242 (7):644–648
(14) Giannitrapani L, Soresi M, La Spada E, Cervello M, D’Alessandro N, Montalto G. Sex hormones and risk of liver tumor. Ann N Y Acad Sci 2006; 1089:228–236.
(15) Jameson JJ, Cave DR. Hormonal and antihormonal therapy for epistaxis in hereditary hemorrhagic telangiectasia. Laryngoscope 2004; 114 (4):705–709.
(16) van Cutsem E, Rutgeerts P, Vantrappen G. Treatment of bleeding gastrointestinal vascular malformations with oestrogen–progesterone. Lancet 1990; 335 (8695):953–955.
(17) Flessa HC, Glueck HI. Hereditary hemorrhagic telangiectasia (Osler-Weber-Rendu disease): management of epistaxis in nine patients using systemic hormone therapy. Arch Otolaryngol 1977; 103 (3):148–151.
(18) McAllister KA, Grogg KM, Johnson DW, Gallione CJ, Baldwin MA, Jackson CE, et al. Endoglin, a TGF-beta binding protein of endothelial cells, is the gene for hereditary haemorrhagic telangiectasia type 1. Nat Genet 1994; 8 (4): 345–351.
(19) Johnson DW, Berg JN, Baldwin MA, Gallione CJ, Marondel I, Yoon SJ et al. Mutations in the activin receptor-like kinase 1 gene in hereditary haemorrhagic telangiectasia type 2. Nat Genet 1996; 13 (2):189-195.
(20) Burt RW, Bishop DT, Lynch HT, Rozen P, Winawer SJ. Risk and surveillance of individuals with heritable factors for colorectal cancer. WHO Collaborating Centre for the Prevention of Colorectal Cancer. Bull World Health Organ 1990; 68 (5): 655–665.
(21) Järvinen HJ, Sipponen P. Gastroduodenal polyps in familial adenomatous and juvenile polyposis. Endoscopy 1986; 18 (6): 230-234.
(22) Lodewijk AA Brosens, Danielle Langeveld, W Arnout van Hattem, Francis M Giardiello, G Johan A Offerhaus. Juvenile polyposis syndrome. World J Gastroenterol 2011 ; 17 (44): 4839-4844.
(23) Desai DC, Neale KF, Talbot IC, Hodgson SV, Phillips RK. Juvenile polyposis. Br J Surg Jan 1995; 82 (1):14–17.
(24) Manfredi M. Hereditary hamartomatous polyposis syndromes: understanding the disease risks as children reach adulthood. Gastroenterol Hepatol (N Y) 2010; 6 (3): 185–196.
(25) Howe JR, Roth S, Ringold JC, Summers RW, Järvinen HJ, Sistonen P et al. Mutations in the SMAD4/DPC4 gene in juvenile polyposis. Science 1998; 280 (5366): 1086–1088.
(26) Howe JR, Bair JL, Sayed MG, Anderson ME, Mitros FA, Petersen GM et al. Germline mutations of the gene encoding bone morphogenetic protein receptor 1A in juvenile polyposis. Nat Genet 2001; 28 (2):184-187.
(27) Jass JR, Williams CB, Bussey HJ, Morson BC. Juvenile polyposis - a precancerous condition. Histopathology 1988; 13 (6): 619-630.
(28) Merg A, Howe JR. Genetic conditions associated with intestinal juvenile polyps. Am J Med Genet C Semin Med Genet 2004; 129C(1): 44–55.
(29) Syngal S, Brand RE, Church JM, Giardiello FM, Hampel HL, Burt RW et al. ACG clinical guideline: genetic testing and management of hereditary gastrointestinal cancer syndromes. Am J Gastroenterol 2015; 110 (2): 223–262.
(30) Cox KL, Frates RC Jr, Wong A, Gandhi G. Hereditary generalized juvenile polyposis associated with pulmonary arteriovenous malformation. Gastroenterology. 1980; 78 (6):1566–15670.
(31) Inoue S, Matsumoto T, Iida M, Hoshika K, Shimizu M, Hisamoto N, et al. Juvenile polyposis occurring in hereditary hemorrhagic telangiectasia. Am J Med Sci 1999; 317 (1): 59–62.
(32) Prieto G, Polanco I, Sarria J, Larrauri J, Lassaletta L. Association of juvenile and adenomatous polyposis with pulmonary arteriovenous malformation and hypertrophic osteoarthropathy. J Pediatr Gastroenterol Nutr 1990; 11 (1): 133–137.
(33) Gallione C, Aylsworth AS, Beis J, Berk T, Bernhardt B, Clark RD et al. Overlapping spectra of SMAD4 mutations in juvenile polyposis (JP) and JP–HHT syndrome. Am J Med Genet Part A 2010; 152A (2):333–339.
(34) Teekakirikul P, Milewicz DM, Miller DT, Lacro RV, Regalado ES, Rosales AM et al. Thoracic aortic disease in two patients with juvenile polyposis syndrome and SMAD4 mutations. Am J Med Genet A 2013; 161A (1):185–191.
(35) Andrabi S, Bekheirnia MR, Robbins-Furman P, Lewis RA, Prior TW, Potocki L. SMAD4 mutation segregating in a family with juvenile polyposis, aortopathy, and mitral valve dysfunction. Am J Med Genet A 2011; 155A (5): 1165–1169.