INTRODUCTION
La maladie de CADASIL est causée par une mutation dans le gène NOTCH 3 sur le bras court du chromosome 19p13.2-p13.1. Le diagnostic repose principalement sur la clinique comportant des migraines, des accidents vasculaires cérébraux, des troubles psychiatriques et des troubles moteurs. Le diagnostic définitif peut être posé soit sur base de l’histopathologie de la biopsie cutanée ou cérébrale, soit quand la clinique et/ou l’imagerie cérébrale sont typiques. Malheureusement, on ne dispose pas encore de traitement curatif ayant fait ses preuves (1).
CAS CLINIQUE
Une femme de 66 ans se présente aux services des urgences suite à une chute dans les escaliers.
À l’anamnèse, la patiente présente des troubles phasiques ainsi qu’un état bradypsychique important. Elle ne se rappelle pas des circonstances exactes de sa chute.
À l’hétéro-anamnèse, son mari trouve que depuis quelques temps elle présente des troubles cognitifs essentiellement des troubles de la mémoire et fait régulièrement des chutes. Parmi ses antécédents, on note principalement un épisode d’aphasie spontanément résolutif en 1981 et un accident vasculaire ischémique dans les territoires de l’artère pariétale antérieure gauche et de l’artère pré-frontale droite avec récupération complète en 1992.
Son traitement journalier consiste en un comprimé d’Adalat et un comprimé d’Asaflow 80 mg. Hormis l’hypertension artérielle, il n’y a pas d’autre facteur de risque vasculaire modifiable (diabète, obésité, tabagisme, hypercholestérolémie). Sur le plan neurologique, il n’y a pas de notion de migraine.
ANAMNÈSE FAMILIALE
Le père de la patiente est décédé suite à une pathologie pulmonaire obstructive, tandis que la mère est morte jeune des suites d’un cancer mammaire.
Un de ses cousins est décédé à 56 ans de cause inconnue et aurait présenté un AVC étant plus jeune.
La patiente est fille unique, elle est mariée et a un fils, âgé de 39ans, ainsi que deux petits enfants (un petit fils de 5 ans et une petite fille de 3 ans).
EXAMEN CLINIQUE
À l’examen neurologique, la patiente est consciente mais tout à fait bradypsychique avec un ralentissement idéatoire important ainsi que des troubles praxiques sévères. Les paires crâniennes sont sans particularité. Il n’y a pas de symptôme latéralisé. Le Barré et le Mingazzini sont tenus symétriquement. Le réflexe cutané plantaire est en extension bilatéralement. Elle présente une discrète attitude hypertonique du membre supérieur droit et une libération des réflexes archaïques frontaux avec présence d’un réflexe de succion et un grasping bilatéral.
Le profil cognitif de la patiente se caractérise par des difficultés exécutives et praxiques à l’avant plan. Le caractère fluctuant des plaintes, associé à de nombreuses chutes, et le tableau cognitif suggèrent une atteinte vasculaire.
Pour le reste, la tension artérielle est de 110/70 mmHg et son rythme cardiaque est régulier. Son auscultation cardio-pulmonaire est normale de même que l’examen abdominal. Elle présente une plaie du cuir chevelu ainsi qu’un hématome avec une plaie suturée au niveau de la joue gauche et un volumineux hématome orbitaire gauche.
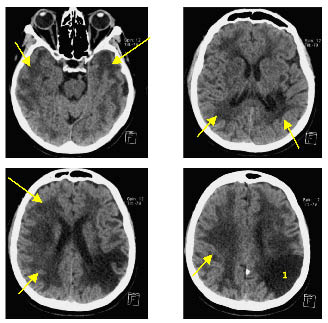
Le scanner cérébral (figure 1) réalisé aux Urgences témoigne d’une atteinte sévère inhabituelle de la substance blanche. Il montre (1) une volumineuse séquelle malacique cortico-sous-corticale pariétale gauche et une autre plus petite temporale gauche avec un aspect fortement hypodense de la substance blanche, évoquant une myélinopathie (flèches). Un bilan neuro-cardio-vasculaire complet et séro-immunitaire est programmé en vue d’exclure les nombreuses maladies vasculaires, infectieuses, auto-immunes potentiellement responsables de ce type d’anomalies …
Figure 1. Scanner cérébral de la patiente
La biologie est tout à fait normale : l’hémogramme, la fonction rénale, l’enzymologie, l’ionogramme et le lipidogramme sont normaux. Elle a un syndrome inflammatoire non significatif avec une CRP à 13.1mg/L (pour une normale < 10mg/L). La fonction thyroïdienne est normale. Le facteur rhumatoïde est légèrement positif (32,5 UI/ml pour une normale < 25 UI/ml) mais aspécifique. Le FAN, les anticorps anti-cardiolipines et les ANCA sont négatifs.
La ponction lombaire est normale avec 1 GB/mm³, une glycorachie (61.1 mg/dL) et une protéinorachie (0.21g/L pour une normale entre 0.1 et 0.45Ig/L) normales ainsi qu’une absence de bande oligoclonale IgG spécifique. La culture ne montre aucune croissance bactérienne.
L’ECG montre un rythme sinusal à 84 bpm.
La radiographie du thorax est banale.
L’IRM cérébrale (figures 2 et 3) ne montre pas de lésion ischémique récente mais (1) une importante séquelle cortico-sous-corticale pariétale gauche et d’autres plus petites lésions en basi-frontale droite et frontale gauche. Il n’y a pas d’hydrocéphalie ni de collection péri-cérébrale. Elle montre également une importante myélopathie étendue sévère de la substance blanche supra-tentorielle atteignant l’ensemble des lobes mais respectant globalement les capsules externes (flèches). Cette myélopathie s’étend également au niveau des fibres en « U ».
Figure 2. Images IRM cérébrale T2 flair et Figure 3. Images IRM cérébrale T2 TSE

L’IRM médullaire ne montre pas d’anomalie de signal ni de morphologie du cordon médullaire, pas de processus expansif intra-canalaire intra ou extra-axial.
À l’angio-IRM cérébrale pas d’occlusion ou de sténose significative au niveau des principales artères intracrâniennes, pas d’anévrysme ni de signe de vasculite.
Le CT-scan thoraco-abdominal n’apporte pas d’indices spécifiques (pas d’adénomégalie, ni de lésion thoraco-abdominale suspecte, ...).
L’EEG démontre un tracé légèrement à modérément perturbé pour l’âge car parfois un peu trop lent et également irrégulier et fluctuant.
L’échotomographie doppler des vaisseaux cervicaux n’objective pas de sténose hémodynamiquement significative.
Le bilan cardiaque, comprenant l’examen échocardiographique réalisé par voie trans-thoracique et par voie trans-œsophagienne ainsi que l’holter-ECG de 24h, est banal.
L’EMG et les potentiels évoqués somesthésiques des membres supérieurs apportent Ides résultats dans les limites de la normale.
Le fond d’œil montre un stade 2 vasculaire mais aucun argument pour une vasculite.
La recherche d’acides gras à très longues chaines, caractéristiques de l’adrénoleucodystrophie, et la recherche d’un déficit enzymatique de l’enzyme alpha-galactosidase, responsable de la maladie de Fabry, sont négatives.
Vu la conjonction symptomatologique (état bradyspsychique, troubles moteurs et cognitifs), les anomalies cérébrales (particulièrement spectaculaires au niveau de la substance blanche) documentées en neuro-imagerie et un bilan étiologique de première ligne non contributif, une maladie de CADASIL est envisagée, nous conduisant à organiser les investigations cytogénétiques signant, d’ordinaire, cette affection rare.
INVESTIGATIONS CYTOGÉNÉTIQUES
L’étude du gène NOTCH3 responsable du syndrome de CADASIL a mis en évidence une substitution en position c.1370 de la Guanine par la Thymine à l’état hétérozygote dans l’exon 8 du gène NOTCH 3. Cette substitution d’une base azotée conduit après transcription ribosomique à la production d’une protéine fille dépourvue d’un résidu cystéine sur un site critique à la conformation 3D de la protéine. Ce changement de la conformation de la protéine est toujours responsable d’une altération fonctionnelle. Le variant identifié est à considérer comme étant la mutation causale très probablement impliquée dans le tableau clinique de notre patiente. La grande majorité des mutations du gène NOTCH 3 impliquées dans le syndrome de CADASIL affecte un résidu de cystéine, les programmes de prédilection des effets de ce variant sur la fonctionnalité de la protéine vont tous dans le sens d’une pathogénicité et il n’est pas décrit dans les bases de données de polymorphismes.
CONCLUSIONS DE LA MISE AU POINT
A posteriori, l’enquête familiale se résume à l’interrogatoire plus poussé de son fils unique. Il signale depuis une dizaine d’années des migraines qui ont justifié une évaluation neurologique en avril 2008.
Une IRM cérébrale, réalisée pour l’occasion, démontrait de multiples lésions en hypersignal T2 et flair de la substance blanche supratentorielle, avec un épais liseré périventriculaire et de multiples lésions réparties au sein de la substance blanche périventriculaire, avec une prédominance bifrontale.
Le tableau clinique et les atteintes objectivées à l’IRM sont à haute suspicion d’être corrélés à la maladie héréditaire présente dans la famille.
La maladie de CADASIL (Cerebral autosomial dominant arteriopathy with subcortical infarcts and leukoencephalopathy) est une angiopathie autosomique dominante héréditaire. Elle fut décrite pour la première fois en 1955 par le neurologue Van Bogaert, à cette époque on parlait de démence héréditaire avec de multiples infarctus. Au début des années 1990, une mutation causale a été mise en évidence dans le gène NOTCH3 sur le bras court du chromosome 19p13.2-p13.1.
La protéine produite par le gène NOTCH3 est cruciale pour la différenciation des cellules musculaires lisses vasculaires ainsi que pour le développement et l’homéostasie des vaisseaux sanguins (1-3).
Le récepteur NOTCH3 est composé d’un large fragment extracellulaire, composé principalement de la répétition de 34 EGF-like peptide, et d’un petit fragment intracellulaire. Les ligands protéiques, principalement les protéines delta et serrate, se fixent sur la partie extracellulaire du récepteur ce qui induit le clivage protéolytique du domaine intracellulaire, lequel entre dans le noyau nucléaire pour modifier l’expression génique.
Une fois que le domaine extracellulaire est fixé à son ligand, une métalloprotéase de la famille ADAM, clive la protéine NOTCH à la limite de la membrane. Cela libère la partie extracellulaire du récepteur NOTCH, qui continue à interagir avec son ligand. Le ligand ainsi que le domaine extracellulaire de NOTCH sont ensuite endocytés par la cellule exprimant le ligand.
Après ce premier clivage, une enzyme appelée γ-sécrétase clive la partie restante de la protéine NOTCH juste à l’intérieur du feuillet interne de la membrane cellulaire de la cellule exprimant NOTCH. Cela libère le domaine intracellulaire de la protéine NOTCH, qui se déplace ensuite vers le noyau où il peut réguler l’expression génique par l’activation du facteur de transcription CSL (4). Lors de son activation, le signal NOTCH entraine une cascade intracellulaire dans la cellule musculaire lisse (figure 4).
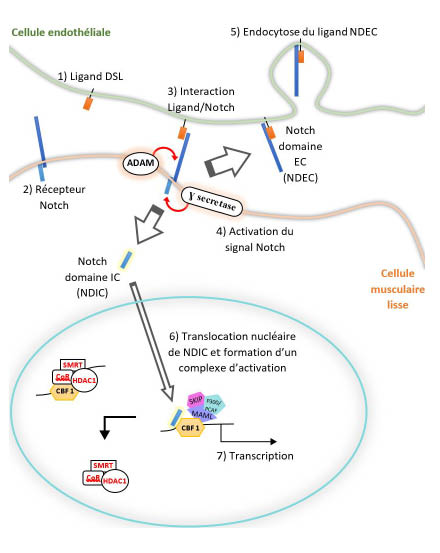
Le signal NOTCH est activé quand un récepteur transmembranaire de la famille du Delta-Serrate LAG (DSL) (1) interagit avec la répétition de peptides EGF-like provenant du domaine extracellulaire des récepteurs NOTCH (2) sur les cellules adjacentes. Les interactions Ligand/récepteur induisent un changement de la conformation du récepteur, exposant des sites critiques pour le clivage des récepteurs NOTCH par les enzymes ADAM17 (S2) et g-secretase (S3) (3). La découpe des récepteurs NOTCH aboutit à la libération et la translocation du domaine intracellulaire (ICD) au noyau (4) tandis que le domaine extracellulaire (ECD) est soumis à l’endocytose par la cellule porteuse de récepteurs (5).
Dans le noyau, le domaine RAM du NOTCH ICD se lie au répresseur de la transcription CBF1, causant le déplacement de corépresseurs associés et l’activation de co-activateurs (6) pour activer la transcription en aval des gènes régulateurs de la production de NOTCH/CBF1 (7) (5).
Figure 4. Vue d’ensemble du signal NOTCH (5)
Suite à cette mutation, le domaine extracellulaire du récepteur NOTCH 3 est incapable de se lier aux protéines delta et serrate, ce qui bloque la voie de signalisation. La protéine NOCTH 3 s’accumule dans les vaisseaux sanguins et, plus précisément, dans la membrane cytoplasmique des cellules musculaires lisses des vaisseaux (6). Cette accumulation progressive, au fil des ans, se traduit par l’apparition, dans le cytoplasme de ces cellules musculaires lisses des micro-vaisseaux concernés, de dépôts granulaires osmiophiliques, donnant en microscopie électronique une image spécifique de la maladie de CADASIL (figure 5). La composition précise de ces dépôts cytoplasmiques demeure à ce stade, inconnue.
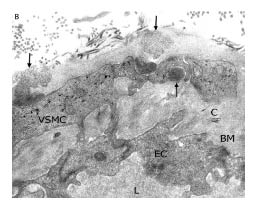
Artérioles montrant des dépots de GOM - matériel granulaire osmiophilique – (flèches) avec des densités électroniques et des tailles diffèrentes, certains d’entre eux montrant des formes bizarres. L – lumière, EC – cellule endothéliale, BM – membrane basale, C – collagène. (7)
Figure 5. Anomalies pathologiques caractéristiques en microscopie électronique dans la maladie de CADASIL (7)
Les quatre symptômes cardinaux de cette pathologie sont :
-
les migraines avec aura ;
-
les accidents vasculaires cérébraux ;
-
les troubles psychiatriques et les troubles de l’humeur ;
-
les troubles cognitifs évoluant vers une démence.
Cette maladie affecte aussi bien les hommes que les femmes. L’âge et l’intensité des symptômes varient d’une personne à l’autre.
Le signe clinique initial, observé chez un quart des patients, est l’apparition de migraines avec aura (visuelle et sensitive) débutant entre 20 et 40 ans. Les accidents vasculaires cérébraux sont observés dans 70 à 80% des cas, débutant habituellement aux alentours de 50 ans. Ils sont typiquement localisés en subcortical.
Les troubles de l’humeur sont observés dans 20% des cas.
Les troubles cognitifs, comme des difficultés de concentration, d’attention et des pertes de mémoire, sont les symptômes les plus précocement observés. Le déclin commence avec l’apparition d’un état apathique, de déficit des fonctions motrices, des difficultés phasiques pour aboutir chez un patient sur 3, après l’âge de 60 ans, à un état de démence. Cet état est souvent associé à d’autres signes de gravité de la maladie comme par exemple des difficultés à la marche, une incontinence urinaire et parfois une paralysie pseudo-bulbaire (1).
La médiane de survie est de 64 ans chez les hommes et de 69 ans chez les femmes (1,8).
Le diagnostic définitif peut être posé soit sur base de l’histopathologie de la biopsie cutanée ou cérébrale, soit quand la clinique et/ou l’imagerie cérébrale sont typiques et en lien avec une mutation dans le gène NOTCH3 (1).
Critères diagnostiques plaidant pour une probable maladie de CADASIL (Davous 1998) (9)
-
Jeune âge de début (< 50 ans)
-
Au moins deux des critères cliniques suivants :
- épisodes d’accidents vasculaires cérébraux avec des signes neurologiques permanents ;
- céphalées migraineuses ;
- troubles majeurs de l’humeur ;
-démence subcorticale. -
Pas de facteur de risque cardiovasculaire lié au déficit
-
Preuve de l’héritage avec une transmission autosomique dominante
-
À l’IRM, anomalies de la substance blanche sans infarctus corticaux
ANOMALIES TYPIQUEMENT OBSERVÉES À L’IRM CÉRÉBRALE
L’IRM montre une anomalie étendue de la substance blanche en l’absence d’infarctus corticaux. Il y a une tendance à une symétrie de l’atteinte. Le centre semi-ovale est atteint, avec une épargne des fibres U. L’atteinte s’étend vers le bas dans la partie externe et extrême de la capsule interne.
Dans les lobes frontaux, l’atteinte s’étend jusqu’aux fibres en U. La substance blanche des lobes temporaux est touchée dans les cas typiques (9, 10).
Il peut y avoir des infarctus lacunaires dans les ganglions de la base, le thalamus, la capsule interne, la substance blanche périventriculaire et le tronc cérébral. Les infarctus corticaux sont rares et de petites tailles (9,10).
Le diagnostic est basé sur la mise en évidence au microscope électronique de matériel granulaire osmiophilique (GOM) adjacent à la membrane des cellules musculaires lisses artériolaires ; la spécificité est de 100% mais la sensibilité est de 50% (7).

Figure 6. Ligne du temps illustrant la période d’apparition des différents symptômes et des signes présents à l’imagerie cérébrale (1)
TRAITEMENT
Les anticoagulants et les agents thrombolytiques, comme la warfarine et l’héparine, ne sont pas préconisés en l’absence de bénéfice et en raison du risque hémorragique. L’aspirine est considérée comme la moins mauvaise alternative (10). Des agents neuroprotecteurs, antioxydants, des antagonistes N-methyl-D-aspartate (NMDA), des antagonistes des canaux Ca voltage dépendant comme la Nimodipine et la Flunarizine, ont aussi été suggérés vu les bénéfices qu’ils semblent avoir dans la prévention des crises migraineuses (11).
Recommandations pratiques
Un diagnostic de maladie de CADASIL doit être évoqué chez un(e) patient(e) jeune souffrant régulièrement de migraines avec aura, ayant présenté un ou plusieurs épisodes d’accident vasculaire cérébral, sans cause clairement établie, et une atteinte importante de la substance blanche, anormale pour l’âge, à l’IRM cérébrale. Aucun traitement n’a actuellement été démontré comme étant bénéfique pour ces patients mais la prise en charge repose essentiellement sur la prévention des AVC, des chutes et des troubles cognitifs ainsi que sur le traitement symptomatique des migraines.
Affiliation
1 Dr. Nathalie Cals, Service de Neurologie, Hôpital de Jolimont
Correspondance
Dr. Emmanuelle Levecque
Cliniques universitaires Saint-Luc
Avenue Hippocrate 10,
B-1200 Bruxelles
E-mail : emmanuelle.levecque@student.uclouvain.be
Références
1. Chabriat H, Bousser MG. Cadasil. EMC 2011 ; 17-046-B-12, 1-13, doi :10.1016/S0246-378(11)52828.
2. Brass S, Smith E, Arboleda-Velasquez J, Copen W, Frosch, M. Case 12-2009: A 46-Year-Old Man with Migraine, Aphasia, and Hemiparesis and Similarly Affected Family Members, N Engl J Med 2009; 360: 1656-65.
6. Baumann M, Junna M, Kalimo H, Miao K, Mykkänen K, Pöyhönen M et al. CADASIL : The most common hereditary subcortical vascular dementia. Future Neurology 2008; 3(6): 683-704.
7. Dziewulska D, Lewandowska E, Parys M, Pasennik E. Ultrastructure of granular osmiophilic material deposits (GOM) in arterioles of CADASIL patients. Folia Neuropathol 2011; 49 (3): 174-180.
9. Van der Knaap MS, Valk J. Magnetic Resonance of Myelination and Myelin Disorders (3rd edition). Springer 2005, Cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy (pp. 541-548).
10. Tikka S, Baumann M, Siitonen M, Pasanen P, Pöyhönen M, Myllykangas L. et al. Pathology & Genetics of (non-CAA) Cerebral Microvascular Disease CADASIL and CARASIL. Brain Patholy ISSN 2014, 24, 525–544, doi:10.1111/bpa
11. Biller, J. Stroke in Children and Young Adults (2nd edition). Saunders Elsevier 2009, 168 – 169.