INTRODUCTION
En postprandial, la glycémie augmente dans le sang et une partie du glucose est stockée sous forme de glycogène dans le foie et les muscles (glycogénogenèse). Le glycogène est un polymère de glucose arborescent, dont les unités sont liées entre elles par des liens osidiques a-1,4 et a-1,6. Lorsque les besoins en glucose augmentent, le stock de glycogène permet de libérer des unités de glucose. Ce mécanisme appelé glycogénolyse fait intervenir plusieurs enzymes dont la phosphorylase kinase. Cette enzyme permet d’hydrolyser les liaisons a-1,4 du glycogène. Un déficit en glycogène phosphorylase ou en glycogène phosphorylase kinase entraine une glycogénose (GSD – Glycogen Storage Disease). Les glycogénoses sont des erreurs innées du métabolisme, majoritairement autosomiques récessives, qui sont caractérisées par l’accumulation anormale de glycogène dans différents tissus (1). Au moins quinze types de glycogénoses ont été identifiés et historiquement, numérotées selon la chronologie de leur description (Tableau 1).
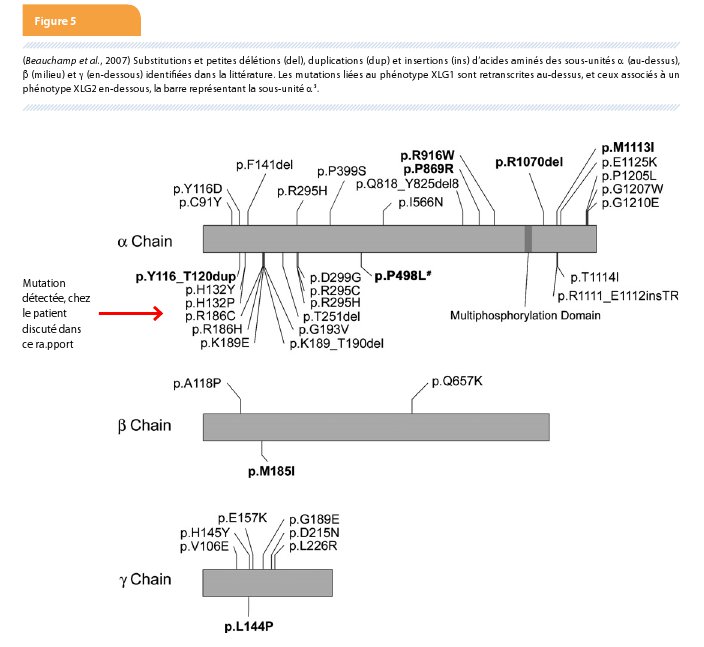
Normalement, la glycogène phosphorylase kinase est activée par phosphorylation et active à son tour la glycogène phosphorylase, qui a pour rôle de scinder les liaisons osidiques a-1,4 du glycogène (2) (Figure 1). En cas de déficit hépatique en glycogène phosphorylase (GSD VI) ou en glycogène phosphorylase kinase (GSD IX), les liaisons a-1,4 du glycogène ne peuvent plus être dégradées. Il y a donc une accumulation du polymère dans le foie, ce qui peut entrainer des symptômes cliniques et des troubles biochimiques. La phosphorylase kinase est constituée de quatre sous-unités : a, ß, g et d. Les sous-unités a et ß ont une fonction régulatrice. L’unité g contient l’activité catalytique et la sous-unité d (ou calmoduline) est, quant à elle, dépendante de la concentration en calcium (3). Ces sous-unités sont codées par différents gènes et il existe également des isoformes spécifiques à certains tissus. Le gène le plus fréquemment muté dans le cas des glycogénoses par atteinte du système phosphorylase – phosphorylase kinase, est le gène PHKA (lié à l’X), qui code pour la sous-unité a (3). De plus, les différentes mutations dans ce gène peuvent être classées en deux sous-types biochimiques de glycogénose liée à l’X (XLG1 et XLG2) ; selon la mesure d’activité de la kinase dans les érythrocytes, normale ou déficitaire (2).

OBSERVATION CLINIQUE
Nous rapportons le cas d’un jeune garçon roumain, âgé de deux ans et huit mois lorsqu’il nous est référé de Roumanie. À l’âge de 20 mois, une hépatomégalie avec augmentation des transaminases est découverte de manière fortuite, suite à un épisode de fièvre et émission de selles acholiques en l’absence d’ictère.
Il s’agit du premier enfant d’un couple de parents non consanguins. Il est né à 38 semaines d’aménorrhée par voie basse. Son poids de naissance était de 3,940 Kg (P 75) et il a présenté une légère détresse respiratoire transitoire suivie par monitoring. Il n’y a pas de notion d’hypoglycémie ou d’ictère néonatal.
L’enfant n’a aucun antécédent médical particulier. Dans ses antécédents familiaux, on note que le papa a souffert d’une hépatite A et que la grand-mère maternelle a quant à elle, présenté une stéatose hépatique avec cholécystectomie.
Au vu de la clinique, la démarche diagnostique a été, dans un premier temps, d’exclure les hépatopathies virales (absence d’immunité anti-HAV et les anti-Hbs à 184 UI/L, prouvant l’immunité vaccinale [réf : < 10 – négatif]) et auto-immunes (Ac anti-LKM1 négatifs, gammaglobulines à 9,5% [réf : 10 – 22]), ainsi que les infections virales à tropisme hépatique (sérologies négatives pour la Toxoplasmose, le Cytomégalovirus et l’Epstein-Barr virus).
Une maladie de Wilson (céruloplasmine à 28 mg/dL [réf : 20 – 60]), un déficit en a-1 antitrypsine (0,97 g/L [réf : 0,9 – 2]), une mucoviscidose (test à la sueur normal, élastase pancréatique dans les selles à 416 µg/g de selles [réf : > 200 – normal]) et une intolérance au gluten (Ac anti-endomysium et HLA DQ2/DQ8 négatifs) ont également été exclus.
D’autres examens ont été effectués en Roumanie. Une échographie hépatique a mis en évidence la présence d’un foie de surcharge et l’élastométrie par Fibroscan® s’est révélée normale (stiffness : 3.8 KPa, IQR : 0.5 KPa). L’électrocardiogramme et l’électroencéphalographie étaient tous deux normaux. Les parents rapportant des épisodes réguliers de fatigue et de perte d’énergie, un profil glycémique a également été effectué chez le patient. Celui-ci s’est avéré normal, mis à part de rares épisodes d’hypoglycémies asymptomatiques (minima à 54,69 mg/dL, après 12 heures de jeûne). Une glycogénose est alors suspectée par les médecins roumains. L’enfant nous est donc envoyé pour une prise en charge, afin d’affiner le diagnostic.
À l’examen clinique, on note un garçon éveillé, avec un développement psychomoteur en relation avec son âge. Il a un poids, une taille et un périmètre crânien qui suivent la courbe du percentile 50.
L’examen cutané est normal. L’abdomen est souple, néanmoins, on constate une importante hépatomégalie à 7-8 cm du rebord costal. Le foie est élastique, lisse et nous n’observons pas de signe d’hypertension portale, ni de circulation collatérale abdominale. Le reste de l’examen clinique cardio-pulmonaire, neurologique et ORL est sans particularités.
D’autres examens complémentaires sont réalisés afin de compléter le bilan déjà effectué en Roumanie.
De nouvelles biologies sanguines montrent des enzymes hépatiques toujours perturbées, oscillant entre 188 et 723 U/L pour les GOT (AST) [réf 15 – 40 U/L], 185 et 673 U/L pour les GPT (ALT) [réf 10 – 40 U/L] et 87 et 401 U/L pour les GGT [réf < 60 U/L]. L’acide lactique est normal à jeun (1,1 mmol/L [réf : 0,5 – 2,2]), mais augmenté en postprandial (5,5 mmol/L) et le pyruvate est également élevé (0,32 mmol/L [réf : 0,03 – 0,10]). L’examen des urines rapporte la présence de corps cétoniques avec un taux élevé d’acide ß-hydroxybutyrate (1599,4 mmol/mol de créatinine [réf : < 5]).
Les sels biliaires, l’ionogramme, l’hémogramme, le lipidogramme, le bilan martial et la fonction thyroïdienne sont quant à eux normaux.
En ce qui concerne les examens hépatiques, l’échographie du foie confirme l’hépatomégalie, ainsi qu’un parenchyme hyperéchogène homogène et une absence de lésion focale. Les contours hépatiques sont discrètement irréguliers en sous-capsulaire, mais il n’y a pas de signe d’hypertension portale. Il n’y a pas d’anomalies de la vésicule, ni de dilatation des voies biliaires.
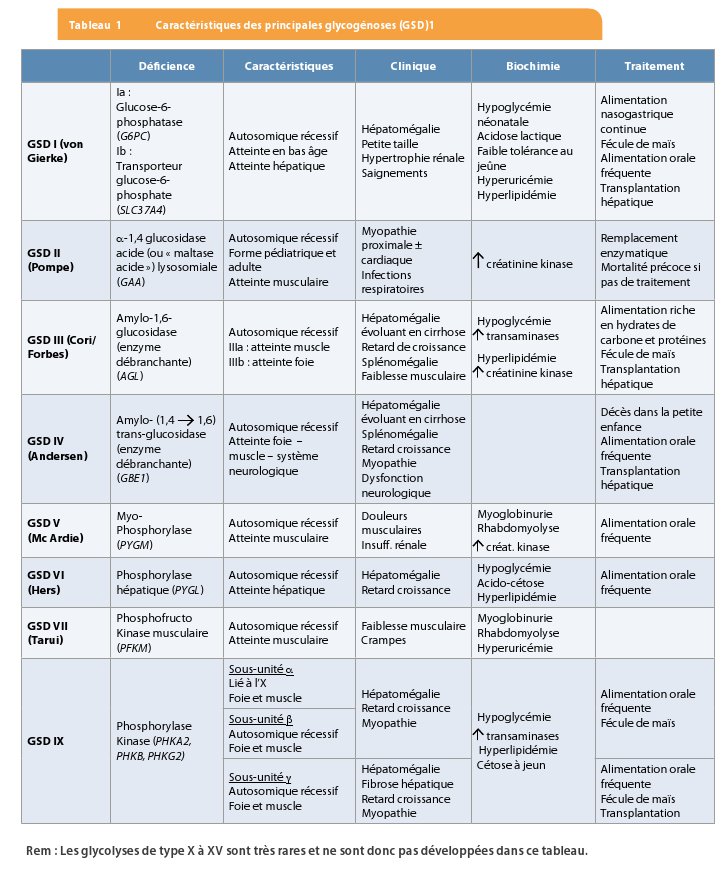
Afin d’affiner la recherche du diagnostic, une biopsie hépatique est effectuée (Figure 2). L’image histologique en coloration de routine (hémalun-éosine) est, selon anatomopathologiste, suggestive d’une glycogénose. Cependant, la stéatose microvésiculaire plaide pour une maladie mitochondriale ou une maladie de surcharge des esters de cholestérols, qui sont donc des diagnostics différentiels à considérer.
L’analyse de l’activité de la lipase acide lysosomiale du patient, nous permet d’exclure le diagnostic de la maladie de stockage des esters du cholestérol (0,74 nmol/punch/h [réf : 0,37 – 2,30]). L’activité de la phosphorylase kinase dans les érythrocytes étant elle aussi normale (13,3 U/g d’hémoglobine [contrôle : 5,5 – 7]), nous excluons également le diagnostic de glycogénose de type IX. Cependant, après discussion multidisciplinaire et étant donné les indices suggestifs d’une glycogénose par atteinte du système de la phosphorylase – phosphorylase kinase (hépatomégalie, bonne tolérance au jeûne, élévation des transaminases et du lactate en postprandial), nous procédons à des analyses génétiques qui mettent en évidence, par l’étude du gène PHKA2, la mutation connue c.556C>T (p. Arg186Cys) (3) à l’état hémizygote. Ceci confirmant, finalement, le diagnostic de glycogénose hépatique de type IX, variant XLG2 étant donné l’activité enzymatique érythrocytaire non diminuée.
DISCUSSION
La clinique du cas précité menait assez naturellement vers une pathologie hépatique ou à répercussions hépatiques. Nous reprenons ici, de manière non exhaustive, les diagnostics différentiels (4) à évoquer devant une situation clinique similaire : hépatites virales et auto-immunes, infections virales de type Toxoplamose, Cytomégalovirus et Epstein-Barr virus, maladie de Wilson, déficit en a-1 antitrypsine, intolérance au gluten, mucoviscidose, intolérance héréditaire au fructose, glycogénoses hépatiques (type I, III, IV, VI et IX), maladies de la chaine respiratoire mitochondriale, maladies de la ß-oxydation des acides gras, maladie de surcharge en esters de cholestérol, certaines mucopolysaccharidoses.
Les équipes médicales ont procédé à de nombreux examens complémentaires afin d’affiner l’orientation du diagnostic et d’en exclure certains.
L’acidose lactique postprandiale et le rapport anatomo-pathologique ont dirigé les médecins vers une pathologie mitochondriale de la chaîne respiratoire (4), plutôt qu’une glycogénose.
Selon une ancienne étude réalisée par Willems et al.(5), les signes cliniques associés à la glycogénose de type IX sont les suivants : hépatomégalie (92%), hypercholestérolémie (76%), hypertriglycéridémie (70%), retard de croissance (68%), transaminases élevées (56%), retard de développement moteur (52%), cétose à jeun (44%).
La présence de lactate élevé n’est pas reprise dans les signes cliniques et biochimiques les plus fréquents de cette étude. Cependant, dans l’étude clinique plus récente de Davit-Spraul et al. (6), comprenant 45 patients suspectés d’avoir une glycogénose VI ou IX, et dans le rapport de 2011 de Goldstein et al.(2), il est rapporté qu’une hyperlactatémie postprandiale a été observée chez la plupart des patients affectés.
Une discussion entre les pédiatres et le laboratoire de biochimie génétique a permis de proposer des pistes de réflexion concernant les signes biochimiques retrouvés dans ce cas de glycogénose liée à une déficience en phosphorylase kinase.
En période de jeûne, le corps utilise le glucose circulant et libère des unités glucose à partir du glycogène. L’insuline diminue et le glucagon augmente, ce qui a pour effet d’activer la glycogénolyse, afin de libérer du glucose et d’éviter les hypoglycémies.
Mais, lorsqu’il n’y a plus de réserves de glycogène ou qu’elles ne peuvent être mises à profit par blocage métabolique, la néoglucogenèse hépatique et la ß-oxydation des acides gras sont des voies métaboliques qui permettent de libérer les substrats énergétiques nécessaires à l’homéostasie.
Dans la glycogénose de type IX (et par extension dans le type VI), la phosphorylase kinase est déficiente et n’active donc pas la phosphorylase catalysant la réaction de phosphorylation du glycogène. Ceci entraine un blocage de la libération des monomères de glucose et l’absence de dégradation du glycogène expose donc le malade aux hypoglycémies, durant les périodes de jeûne ou de besoin catabolique accru comme un effort physique ou une infection par exemple.
En postprandial, l’insuline augmente et permet de mobiliser le glucose dans les hépatocytes et de maintenir une glycémie constante. La glycogénogenèse est alors activée, pour stocker les unités de glucose. Cependant, les réserves de glycogène du patient souffrant d’une glycogénose IX (et VI) sont saturées, puisque le glycogène n’est pas catabolisé durant les périodes de jeûne. Une partie du glucose en circulation ne peut donc pas être stocké et celui-ci est alors consommé par la glycolyse, formant du pyruvate et du lactate (Figure 3). Il est à noter que d’autres voies métaboliques peuvent dégrader le glucose ne pouvant se lier au glycogène (Figure 4) :
- Voie des pentoses phosphates produisant de l’acide urique
- Production d’acides aminés à partir du pyruvate
- Entrée de l’acétyl-CoA dans le cycle de Krebs
- ß-oxydation produisant des corps cétoniques
- Lipogenèse et synthèse de cholestérol

L’équipe médicale avait exclu la déficience en phosphorylase kinase, puisque l’étude de l’activité érythrocytaire de celle-ci s’était révélée normale. Cependant, la discussion avec le laboratoire de biochimie génétique et une revue de la littérature scientifique a montré qu’il existe un sous-type de glycogénose de type IX lié à l’X dont l’activité enzymatique érythrocytaire reste normale (2,6,9). En effet, dans le gène PHKA2 (lié au chromosome X), une cinquantaine de mutations pathologiques ont été identifiées (2). Celles-ci sont divisées en deux sous-types biochimiques, selon l’activité enzymatique dans les différents tissus. Dans le sous-type 1 (X-linked glycogenosis 1 – XLG1), l’activité de la phosphorylase kinase est déficiente dans les hépatocytes et dans les érythrocytes. Dans le sous-type 2 (XLG2), qui est plus rare, l’activité de la phosphorylase kinase est variable dans les hépatocytes, mais normale voire élevée dans les globules rouges, ce qui représente un piège pour le clinicien.
C’est pourquoi, une activité normale de la phosphorylase kinase érythrocytaire, ne permet pas d’exclure formellement le diagnostic de glycogénose IX. Le séquençage du gène PHKA2 a mis en évidence la mutation c.556C>T (p.Arg186Cys) à l’état hémizygote ; alors que l’activité de la phosphorylase kinase érythrocytaire était normale. Le diagnostic de glycogénose de type IX a donc pu être fait à ce moment. Cette mutation a déjà été rapportée et fait partie du sous-type XLG2 des glycogénoses de type IX liées à l’X (Figure 5).
En ce qui concerne le traitement, il n’existe pas de médication spécifique pour la glycogénose de type IX et celui-ci sera uniquement symptomatique. Les épisodes d’hypoglycémie seront évités par la prise de repas fréquents, riches en hydrates de carbone et en protéines. Il faut également éviter les périodes de jeûne prolongé, les grandes quantités de sucres simples et il est conseillé de prendre du dextrose avant un exercice physique intense. La prise de fécule de maïs (à raison de 1g/Kg) avant le coucher peut être nécessaire afin d’empêcher les hypoglycémies nocturnes et d’optimaliser la croissance staturo-pondérale. Cependant, ces mesures diététiques pourront être abandonnées selon l’évolution de la clinique, puisque les symptômes de la glycogénose de type IX liée à l’X, ont tendance à s’améliorer avec l’âge adulte.
Il est également recommandé de suivre régulièrement l’évolution de la glycémie ainsi que la cétonémie, et enfin, suivre l’évolution par échographie du foie surchargé (2).
CONCLUSIONS
Les glycogénoses sont des maladies métaboliques rares dont le diagnostic n’est pas toujours évident. La déficience en phosphorylase kinase, est la glycogénose la plus fréquente (10). Elle est généralement évoquée devant une hépatomégalie, une élévation des transaminases hépatiques et du lactate postprandial avec une tendance aux hypoglycémies malgré un jeûne assez bien toléré.
Dans la majorité des cas de glycogénose de type IX lié à l’X (gène PHKA2), les symptômes sont modérés et d’évolution favorable, mais aucune corrélation génotype – phénotype n’a pu être démontrée jusqu’à présent. De plus, il a été rapporté que les patients ayant les mêmes mutations peuvent avoir des symptômes cliniques différents (2).
Recommandations pratiques
• Penser à une glycogénose de type IX lorsqu’un patient se présente avec une hépatomégalie, une élévation des transaminases hépatiques et une hyperlactatémie postprandiale. Une tendance aux hypoglycémies peut être observée grâce à un profil glycémique, mais le jeûne est généralement assez bien toléré.
• Même si l’activité de la phosphorylase kinase est normale dans les érythrocytes, il est utile de procéder à une analyse génétique des gènes PHKA2 et PYGL afin d’exclure formellement une glycogénose de type IX, chez les garçons.
Affiliations
a Médecin Assistant Candidat Clinicien Spécialiste (MACCS) en pédiatrie, UCL, Belgique
b Médecin Assistant Candidat Clinicien Spécialiste (MACCS) en Biologie Clinique - Laboratoire de Biochimie Génétique, Cliniques universitaires Saint-Luc, Bruxelles, Belgique
c Service d’Anatomie pathologique, Cliniques universitaires Saint-Luc, Bruxelles, Belgique
d Service de Gastro-Entérologie Pédiatrique, Cliniques universitaires Saint-Luc, Bruxelles, Belgique
Centre d'Investigation Clinique Pédiatrique, Cliniques Universitaires Saint-Luc, Bruxelles, Belgique, Institut de Recherche Expérimentale et Clinique –IREC, UCL, Belgique
Correspondance
Dr. Coralie De Bruyne
Cliniques universitaires Saint-Luc
Pédiatrie
B-1200 Bruxelles
coralie.debruyne@student.uclouvain.be
Références
1. Hendriksz CJ, Gissen P. Glycogen Storage disease. Paediatrics and Child Health 2010; 21(2): 84-89.
2. Goldstein J, Austin S, Kishnani P, Bali, D. Phosphorylase Kinase Deficiency. Gene Reviews - National Center for Biotechnology Information (NCBI) Bookshelf 2011.
En ligne http://www.ncbi.nlm.nih.gov/books/NBK55061/
3. Beauchamp NJ, Dalton A, Ramaswami U, Niinikoski H, Mention K, Kenny P, et al. Glycogen Storage disease type IX : High variability in clinical phenotype. Mol Genet Metab 2007; 92: 88-99. doi:10.1016/j.ymgme.2007.06.007
ouvrir dans Pubmed
4. Stephenne X, Sokal E. Les maladies hépatiques rares de l’enfant. En ligne http://www.pediatrie.be/fr/les-maladies-hepatiques-rares-de-l_enfant/1183/2
5. Willems PJ, Gerver WJ, Berger R, Fernandez J. The natural history of liver glycogenosis due to phosphorylase kinase deficiency: a longitudinal study of 41 patients. EurJ Paediat 1990; 149(4): 268-271.
ouvrir dans Pubmed
6. Davit-Spraul A, Piraud M, Dobbelaere D, Valayannopoulos V, Labrune P, Habe D, et al. Liver glycogen Storage diseases due to phosphorylase system deficiencies : Diagnosis thanks to non invasive blood enzymatic and molecular studies. Mol Gen Metab 2011; 104: 137-143. doi:10.1016/j.ymgme.2011.05.010
ouvrir dans Pubmed
7. Sarafoglou K, Hoffmann GF, Roth KS. Pediatric endocrinology and inborn errors of metabolism. McGraw-Hill Medical, 2009.
8. Rawn JD. Traité de biochimie. De Boeck Université, 1990.
9. Roscher A, Patel J, Hewson S, Nagy L, Feigenbaum A, Kronick J, et al. The natural history of glycogen Storage disease types VI and IX : long-term outcome from the largest metabolic center in Canada. Mol Gen Metab 2014; 113: 171-176. doi:10.1016/j.ymgme.2014.09.005
ouvrir dans Pubmed
10. Bali DS, Goldstein J, Fredrickson K, Rehder C, Boney A, Austin S, et al. Variability of disease Spectrum in children with liver phosphorylase kinase defiency caused by mutations in the PHG2 gene. Mol Gen Metab 2014; 111: 309- 313. doi:10.1016/j.ymgme.2013.12.008
ouvrir dans Pubmed