La protéine tau, physiologiquement présente dans les neurones, stabilise les microtubules de leur cytosquelette axonal. Cependant, il importe également de conserver une flexibilité de ce cytosquelette afin d’assurer des capacités d’apprentissage tout au long de la vie. Deux processus sont impliqués dans cet équilibre entre stabilité et flexibilité des connexions neuronales (Figure 1a) : (a) la proportion variable des isoformes de la protéine tau possédant trois (tau 3R) ou quatre (tau 4R) domaines se liant aux microtubules ; par exemple, le fœtus n’exprime que des isoformes tau 3R, privilégiant ainsi la flexibilité à la stabilité ; (b) la protéine tau va être modifiée, notamment phosphorylée, après sa traduction, ce qui va la détacher des microtubules ; l’équilibre entre phosphorylation et déphosphorylation de cette protéine permet de maintenir les connexions neuronales importantes, et de réorganiser celles qui le nécessitent lors d’apprentissages (1).
Les tauopathies représentent une vingtaine de maladies neurodégénératives caractérisées par l’accumulation anormale de la protéine tau sous forme d’agrégats insolubles à l’intérieur des neurones et des cellules gliales du cerveau. Ces agrégats entraînent la mort des neurones atteints, aboutissant à un déclin cognitif irréversible. Bien qu’elles aient en commun l’agrégation de la protéine tau, les tauopathies diffèrent considérablement quant aux caractéristiques des agrégats (conformation, localisation tissulaire et subcellulaire, etc..) et des régions cérébrales où la maladie prend naissance (1). En fonction des régions cérébrales touchées, les symptômes cliniques varient, et vont souvent se superposer aux stades tardifs de la maladie, ne permettant que difficilement de différencier ces tauopathies du vivant des personnes qui en sont atteintes.
Le traitement des tauopathies nécessiterait des médicaments empêchant l’agrégation de la protéine tau, ou qui induiraient l’élimination des protéines tau anormales. Or, les mécanismes biochimiques à l’origine de l’agrégation de la protéine tau ne sont toujours pas élucidés (2). Néanmoins, ils sont vraisemblablement divers, car la conformation de la protéine dans les agrégats diffère selon la tauopathie (3). Les médicaments ciblant un mécanisme hypothétique d’agrégation devront être administrés à des patients dont la maladie est déterminée avec précision, une condition difficile à remplir pour les tauopathies dites primaires (i.e., autres que celle de la maladie d’Alzheimer) en raison de l’absence actuelle de biomarqueurs spécifiques. Le succès des essais thérapeutiques dépend donc d’un diagnostic différentiel non-biaisé des tauopathies, à l'échelle moléculaire et in vivo, ce qui permettrait de classifier les patients de manière appropriée et de déterminer l’efficacité biologique de chaque médicament testé.
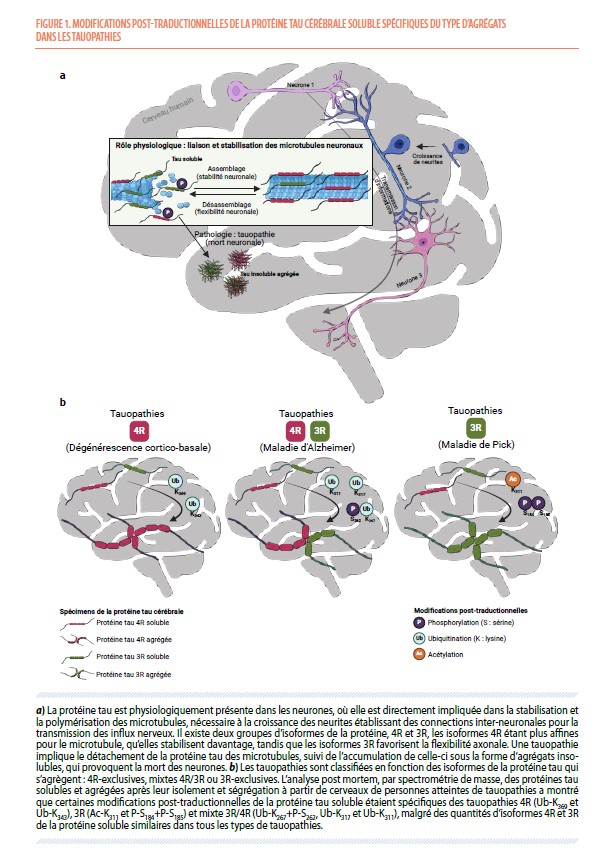
La maladie d’Alzheimer peut être diagnostiquée in vivo par la détection d’une diminution de la concentration en peptide amyloïde β et d’une augmentation des concentrations en protéine tau et en protéine tau phosphorylée sur son résidu thréonine en position 181 (P-T181) dans le liquide cérébrospinal. Dans les autres tauopathies, l’augmentation des concentrations en protéine tau dans le liquide cérébrospinal est variable (4), et le diagnostic final repose actuellement sur l’examen neuropathologique du cerveau post mortem. À l’autopsie, les tauopathies sont classifiées en fonction des isoformes de la protéine tau présentes dans les agrégats. En effet, le cerveau humain adulte présente six isoformes différentes de cette protéine, dont trois sont qualifiées de « tau-3R » et trois autres de « tau-4R » (5) selon le nombre de répétitions du domaine de liaison aux microtubules. Dans la maladie d’Alzheimer, les agrégats sont constitués d’isoformes 3R et 4R : elle est donc classifiée comme tauopathie 3R/4R. La dégénérescence cortico-basale et la paralysie supra-nucléaire progressive sont des tauopathies 4R, tandis que la maladie de Pick est une tauopathie 3R. Enfin, dans les démences fronto-temporales dues à la protéine tau, les agrégats sont le plus souvent soit 3R, soit 4R (6).
Plusieurs chercheurs ont tenté de différencier les tauopathies par la mesure des isoformes 3R et 4R de la protéine tau dans le liquide cérébrospinal, mais cette stratégie s’est avérée inopérante (7, 8). De plus, depuis la découverte de la protéine tau, les seuls travaux ayant mis en évidence des différences moléculaires entre les tauopathies ont été réalisés sur la protéine tau insoluble (i.e., sur les agrégats) provenant du cerveau des personnes après leur décès. En faisant l’hypothèse que ce sont les caractéristiques moléculaires de la protéine tau cérébrale soluble, et non des agrégats, qui représenteraient le mieux celles de la protéine tau dans le liquide cérébrospinal, et que les processus pathologiques conduisant à l’agrégation sont à rechercher sur la protéine soluble, nous avons analysé des échantillons de tissu cérébral prélevés post mortem sur des sujets témoins et des sujets atteints de tauopathie mixte 3R/4R (maladie d’Alzheimer), 3R (maladie de Pick), 4R (dégénérescence cortico-basale), ou de démence fronto-temporale due à la protéine tau, en utilisant la chromatographie en phase liquide couplée à la spectrométrie de masse en tandem (LC-MS/MS) pour la distinction des protéines tau soluble et insoluble. Après ségrégation des protéines solubles et insolubles, ces fractions ont été analysées par spectrométrie de masse, et nous y avons déterminé les quantités des isoformes 3R et 4R, et les modifications post-traductionnelles de la protéine tau (9).
Cette analyse a d’abord montré que la différence de quantités des isoformes 3R et 4R de la protéine tau dans les agrégats selon les tauopathies, que nous avons confirmée, n’existe pas pour la protéine soluble (Figure 1b). Pour celle-ci, il n’y avait pas de différence significative entre les tauopathies, ni avec le groupe témoin, ce qui explique l’échec des tentatives précédentes de différenciation 3R/4R des tauopathies par l’analyse du liquide cérébrospinal (qui ne contient pas d’agrégats de la protéine). Nous nous sommes alors intéressés aux modifications post-traductionnelles de la protéine tau, principalement dans la fraction soluble, afin de déterminer si elles pouvaient permettre de prédire le type d’isoformes retrouvées dans les agrégats, et donc de caractériser la maladie. Nous avons ainsi mis en évidence l’existence de modifications post-traductionnelles de la protéine tau soluble spécifiques des tauopathies 4R (ubiquitination sur les résidus lysine 343 [Ub-K343] et 369 [Ub-K369]) et des tauopathies 3R (acétylation sur le résidu lysine 311 [Ac-K311] et double-phosphorylation sur les résidus sérine 184 et 185 [P-S184+P-S185]). Cette analyse a également révélé qu’il existe des modifications post-traductionnelles de la protéine tau soluble spécifiques de la maladie d’Alzheimer, et qu’on ne retrouve pas dans d’autres tauopathies (Figure 2a), contrairement à d'autres modifications post-traductionnelles récemment utilisées comme biomarqueurs de cette maladie (P-T217, P-T231, P-S396), qui permettent effectivement de distinguer les personnes atteintes de la maladie des personnes qui ne le sont pas, mais pas des personnes atteintes d’autres tauopathies (Figure 2b). Toutes ces modifications post-traductionnelles de la protéine tau nouvellement identifiées constituent donc autant de marqueurs moléculaires à analyser dans le liquide cérébrospinal des patients pour un diagnostic différentiel in vivo des tauopathies, incluant la maladie d’Alzheimer.

Références
- Arendt T, Stieler JT, Holzer M. Tau and tauopathies. Brain Res Bull. 2016 ; 126 : 238-92.
- Seidler PM, Boyer DR, Rodriguez JA, et al. Structure-based inhibitors of tau aggregation. Nat Chem. 2018 ; 10 : 170-6.
- Shi Y, Zhang W, Yang Y, et al. Structure-based classification of tauopathies. Nature. 2021 ; 598 : 359-63.
- Skillback T, Farahmand BY, Rosen C, et al. Cerebrospinal fluid tau and amyloid-b1-42 in patients with dementia. Brain. 2015 ; 138 : 2716-31.
- Lebouvier T, Pasquier F, Buee L. Update on tauopathies. Curr Opin Neurol. 2017 ; 30 : 589-98.
- Spillantini MG, Goedert M. Tau protein pathology in neurodegenerative diseases. Trends Neurosci. 1998 ; 21 : 428-33.
- Luk C, Compta Y, Magdalinou N, et al. Development and assessment of sensitive immuno-PCR assays for the quantification of cerebrospinal fluid three- and four-repeat tau isoforms in tauopathies. J Neurochem. 2012 ; 123 : 396-405.
- Barthelemy NR, Fenaille F, Hirtz C, et al. Tau protein quantification in human cerebrospinal fluid by targeted mass spectrometry at high sequence coverage provides insights into its primary structure heterogeneity. J Proteome Res. 2016 ; 15 : 667-76.
- Kyalu Ngoie Zola N, Balty C, Pyr Dit Ruys S, et al. Specific post-translational modifications of soluble tau protein distinguishes Alzheimer’s disease and primary tauopathies. Nat Commun. 2023 ; 14 : 3706.
Affiliations
1 Université catholique de Louvain (UCLouvain), Institut de neurosciences (IONS), Bruxelles, Belgique.
2 Université catholique de Louvain (UCLouvain), Institut de Duve (DDUV), Phosphorylation des protéines (PHOS), Bruxelles, Belgique.
3 Université catholique de Louvain (UCLouvain), Institut de Duve (DDUV), Plateforme MASSPROT, Bruxelles, Belgique.
4 Cliniques universitaires Saint-Luc, Département de neurologie, Bruxelles, Belgique.
5 Grand Hôpital de Charleroi, Charleroi, Belgique.
* Ces auteurs ont contribué conjointement à la publication
Correspondance
Pr Bernard Hanseeuw
Université catholique de Louvain (UCLouvain), Institut de neurosciences (IONS)
Cliniques universitaires Saint-Luc
Département de neurologie
Avenue Hippocrate 10
B-1200 Bruxelles
bernard.hanseeuw@saintluc.uclouvain.be