INTRODUCTION
L’hyperplasie macronodulaire bilatérale des surrénales (HMBS) est une cause rare de syndrome de Cushing (1). Elle est souvent diagnostiquée vers 50-60 ans de manière fortuite à l’occasion d’une imagerie abdominale (1). Sa distribution est égale entre les sexes (1). Nous rapportons le cas d’un patient présentant des incidentalomes surrénaliens bilatéraux chez qui nous mettons en évidence un hypercorticisme frustre secondaire à la présence de récepteurs illégitimes pour la vasopressine et la sérotonine.
CAS CLINIQUE
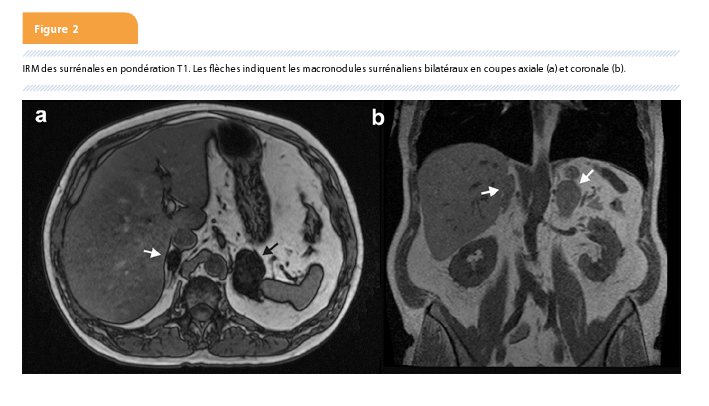
Un patient de 62 ans est adressé au service d’endocrinologie pour bilan de masses surrénaliennes découvertes fortuitement lors d’un scanner abdominal réalisé lors d’une mise au point d’hémochésies non douloureuses. Les examens gastro-entérologiques avaient conclu à un saignement dont l’origine pouvait être un polype rectal, une congestion hémorroïdaire ou un diverticule. Ses antécédents personnels et familiaux sont sans particularité. Il ne prend aucun traitement. Ses seuls symptômes comportent de la fatigue et une gêne lombaire bilatérale d’allure mécanique. Il n’a pas de malaises hypertensifs. L’examen clinique révèle une obésité de grade 1 (IMC 30 kg/m²) et une tension artérielle à 160/90 mmHg. Il ne décèle pas de visage lunaire ni d’ecchymose ni de signe de myopathie des membres inférieurs. À la biologie, l’HbA1c est à 6.2%, témoignant d’un prédiabète, l’aldostérone est normale (0.16 nmol [VN : 0.08-0.4]), le DHEAs est infranormal (0.7 µM [VN : 2.5-6]), le cortisol de 16 h est à 10.3 µg/dl et l’ACTH est abaissé (2 pg/ml [VN : 5-49]). La cortisolurie de 24 h est normale (17 µg/24h [VN <60]) mais le freinage du cortisol au test de suppression nocturne à la dexaméthasone 1 mg (TSD 1mg) est insuffisante (6.8 µg/dl [NV < 1.8]). La vérification du scanner confirme un nodule surrénalien droit de 25x18x31 mm et un gauche de 35x49x39 mm (Figure 1). Leur densité à blanc est inférieure à 10 unités Hounsfield. La résonance magnétique confirme leur nature adénomateuse sur base d’une chute du signal supérieure à 20% (Figure 2). Les catécholamines urinaires de 24 h montre une élévation de la noradrénaline (119 µg/24h [VN <97]), des normétanéphrines (861 µg/24h [VN <390]) et de l’acide homovanilique (8.3 mg/24h [VN <6.6]). L’adrénaline, les métanéphrines, la dopamine et l’acide vanilmandélique sont normaux.

Une scintigraphie à la métaiodobenzylguanidine (MIBG) est réalisée mais ne révèle pas de fixation anormale du traceur, permettant d’écarter un phéochromocytome. Le scanner des surrénales recontrôlé six mois plus tard montre une stabilité des nodules. Nous décidons de poursuivre le bilan vu l’hypercorticisme frustre (ACTH bas, DHEAs bas, freinage insuffisant du cortisol au test de suppression à la dexaméthasone 1 mg), l’obésité, l’HTA et le prédiabète. Un test long à la dexaméthasone faible dose (test de Liddle, 0.5 mg/6h pendant 48h) est réalisé. Le cortisol dosé au jour 3 est à 7.5 µg/dl (VN <1.8) et la cortisolurie collectée du jour 2 au jour 3 est à 15 µg/24 h (VN <10), confirmant l’hypercorticisme. Devant cet hypercorticisme frustre ACTH indépendant associé à une hyperplasie macronodulaire bilatérale des surrénales, nous recherchons la présence de récepteurs illégitimes. Les différents tests sont réalisés selon le protocole établi par A. Lacroix (1). Ce bilan comprend un test d’orthostatisme, un repas mixte et divers tests pharmacologiques : Synacthen®, LH-RH, TRH, glucagon, glypressine et métoclopramide. Aucune élévation du cortisol n’est notée durant le test d’orthostatisme, le repas mixte et les tests à la LH-RH, TRH et glucagon, permettant d’exclure des récepteurs illégitimes à l’angiotensine 2, aux catécholamines, au GIP, au GnRH, LH, FSH, à la TRH, TSH, prolactine et au glucagon, respectivement. La réponse de la 17 hydroxy-progestérone au test à l’ACTH permet d’écarter une forme tardive d’hyperplasie congénitale des surrénales. On note par contre une élévation anormale du cortisol à l’injection de glypressine et à la prise orale de métoclopramide suggérant ainsi la présence de récepteurs illégitimes à la vasopressine et à la sérotonine, respectivement (Figure 3). À l’heure actuelle, il n’existe pas de thérapie ciblée pour bloquer ces récepteurs illégitimes et le seul traitement disponible lorsqu’il s’impose est la surrénalectomie unilatérale voire bilatérale. Un choix chirurgical se fait essentiellement en fonction des complications liées à l’hypercorticisme. Chez notre patient, nous avons opté pour un suivi compte tenu de l’absence de diabète à l’hyperglycémie orale provoquée, d’une tension artérielle bien contrôlée et une densitométrie osseuse normale.

DISCUSSION
Les incidentalomes surrénaliens sont rapportés dans 5% de la population (2-4) et sont bilatéraux dans 10-15% (5). Ils sont responsables d’un syndrome de Cushing dans 35% des cas (4). L’HMBS est une cause rare de syndrome de Cushing (<1%) qui est le plus souvent subclinique (1). En condition physiologique, la sécrétion de cortisol est régulée par l’ACTH via son récepteur spécifique de la mélanocortine de type 2, un récepteur couplé à une protéine G (RCPG), exprimé dans la zone fasciculée. En se fixant sur ce récepteur, l’ACTH induit une cascade enzymatique à l’origine de la production du cortisol. Dans l’HMBS, le cortisol est principalement régulé par des RCPG illégitimes. Ceux-ci peuvent ainsi induire une stéroïdogenèse en réponse à diverses hormones telles que le GIP, les catécholamines, la vasopressine, le glucagon, la sérotonine, l’angiotensine 2, la LH ou l’HCG (2). Parmi ceux-ci, les récepteurs illégitimes pour la vasopressine et la sérotonine sont les plus fréquents (6-8), comme illustré dans ce cas. Dans l’HMBS, l’ACTH est habituellement freiné en raison de l’hypercortisolisme. Il n’est cependant pas correct de parler de syndrome de Cushing ACTH indépendant car il a été démontré récemment une production ectopique d’ACTH par les cellules surrénaliennes (9). Dans l’HMBS avec syndrome de Cushing, 80% des patients ont un récepteur illégitime et 55% des patients en ont plusieurs (6-8). Ce sont ces récepteurs qui sont à l’origine de la formation de macronodules surrénaliens (1).
La présence de récepteurs illégitimes peut être mise en évidence au moyen de tests pharmacologiques selon le protocole de détection proposé par A. Lacroix (1). Les tests sont réalisés sur une période de trois jours après une nuit de jeûne. Au préalable, une suppression à la dexaméthasone 1mg/6h pendant 48h est réalisée afin de supprimer l’ACTH et écarter un éventuel effet de celui-ci sur la stéroïdogenèse (1). Voici les divers types de récepteurs illégitimes et les méthodes pour les détecter :
- Le récepteur illégitime au GIP ou syndrome de Cushing alimentaire. Le GIP est sécrété en réponse à une charge orale de lipide et de glucose (et non lors d’une injection du glucose IV) (1). La présence de récepteur illégitime pour le GIP peut donc être recherchée par un repas mixte (2). S’il est présent, le cortisol augmente en postprandial en réponse à une sécrétion physiologique de GIP alors que le cortisol à jeun est bas en raison de la suppression de l’ACTH. En revanche, s’il coexiste d’autres récepteurs illégitimes, le cortisol à jeun peut être élevé.
- Le récepteur illégitime à la vasopressine. Les récepteurs V2 et V3 sont exprimés de manière aberrantes contrairement au récepteur V1 qui peut être exprimé normalement dans le cortex surrénalien (10). Il serait donc plus correct de parler d’une réponse aberrante ou exagérée du cortisol selon le type de récepteur à la vasopressine (1). Le test consiste en l’injection de glypressine 0.5 mg IV mais il peut aussi être évalué par le test de posture qui stimule la sécrétion de vasopressine. Le type de récepteur ne peut être décelé sur base de ce test.
- Le récepteur illégitime aux catécholamines. L’élévation des catécholamines endogènes induit dans ce cas une élévation pathologique du cortisol dû à la présence de récepteurs β adrénergiques ectopiques. Ils peuvent être mis en évidence par le test de posture (1).
- Le récepteur illégitime à la LH/HCG. L’hypercortisolisme est présent typiquement pendant la grossesse ou en post-ménopause et peut être évalué par le test à la LH-RH (1).
- Le récepteur illégitime à la sérotonine peut être révélé par le métoclopramide qui est un agoniste du récepteur 5HT4 (1).
- Le récepteur illégitime à l’angiotensine peut être évalué par un test de posture (1).
Le diagnostic est important car l’hypercorticisme même frustre augmente la mortalité. Les principales causes de décès sont d’origines cardiovasculaire et infectieuse. Quel que soit l’étiologie du SC, la mortalité est multipliée par un facteur 2 l’année du diagnostic et reste plus élevée après traitement pour des suivis de 12 ans en moyenne par rapport aux contrôles (11). La qualité de vie est également altérée chez les patients en rémission de leur syndrome de Cushing (12). Le diagnostic du syndrome de Cushing subclinique est parfois difficile et les critères diagnostiques diffèrent selon les études (3, 13) : absence de signes cliniques de syndrome de Cushing, suppression insuffisante du cortisol au TSD 1 mg (> 1.8 µg/dl), cortisolurie de 24 h normale, cortisol sérique ou salivaire de minuit légèrement élevé, ACTH partiellement supprimé. Le DHEAs est souvent freiné. Des études ont montré un excès de morbi-mortalité en particulier cardio-vasculaire chez les patients avec SC subclinique comparé aux patients présentant un adénome surrénalien non fonctionnel (14-16). Le taux de cortisol après TSD 1 mg apparaît comme un facteur indépendant de mortalité toute cause confondue (15).
Le traitement des HMBS dépend principalement de la sévérité de l’hypercortisolisme et de ses complications. La présence d’une hypertension artérielle, d’un diabète, d’une ostéoporose ou de manifestations neuropsychologiques est à prendre en considération. Dans les syndromes de Cushing sévères (cortisolurie > 2x la limite supérieure de la normale), la surrénalectomie bilatérale est souvent nécessaire. Dans les cas moins sévères (cortisolurie < 2x la limite supérieure de la normale), on privilégie une surrénalectomie unilatérale portant sur la surrénale la plus volumineuse ou sur la glande montrant une fixation plus élevée de l’iodocholestérol marqué (17). Les inhibiteurs de la stéroïdogenèse comme le kétoconazole ou la metyrapone peuvent aussi être envisagés. Certains récepteurs illégitimes peuvent faire l’objet de thérapies ciblées comme les analogues de la somatostatine dans le syndrome de Cushing alimentaire (2). Mais malheureusement, la réponse au traitement s’épuise rapidement quel que soit l’analogue de la somatostatine utilisé (y compris le pasiréotide) (18). Les β bloquants peuvent être utilisés dans le HMBS avec test de posture pathologique (2). Pour les autres récepteurs illégitimes, il n’existe pas à ce jour de thérapies ciblées. Un suivi annuel par biologie et scanner est recommandé dans les cas non chirurgicaux (1). Le choix thérapeutique n’est donc pas sans conséquence car il met en balance les complications du syndrome de Cushing et celles d’une surrénalectomie bilatérale. La surrénalectomie bilatérale dans le SC (toute cause confondue) est responsable d’une crise d’insuffisance surrénalienne aigue chez 9.3/100 patients-année et d’un taux de mortalité médian de 17% (19). Des études récentes ont montré que les formes sporadiques étaient moins fréquentes qu’on ne le pensait. La mutation du gène ARMC5 sur la tumeur est retrouvée dans plus de 50% des cas des HMBS (4, 20), en particulier chez des patients présentant une réponse aberrante au test de posture, à la vasopressine et au métoclopramide (2). Un screening familial est par conséquent recommandé au moyen d’un test de suppression nocturne à la dexaméthasone 1 mg (1), ce test étant plus sensible que la cortisolurie de 24 h dans le bilan des incidentalomes surrénaliens (21).
CONCLUSION
Nous rapportons le cas d’une HMBS de découverte fortuite compliquée d’un syndrome de Cushing subclinique dû à la présence de récepteurs illégitimes pour la vasopressine et la sérotonine.
Recommandations pratiques
Devant des macronodules surrénaliens, pensez à rechercher un syndrome de Cushing en réalisant un test de suppression nocturne à la dexaméthasone 1 mg. En cas de confirmation de l’hypercorticisme, recherchez la présence de récepteurs illégitimes.
Affiliations
(1) Service d’Endocrinologie, CHU UCL Namur
Correspondance
Dr. Corinne Jonas
CHU UCL Namur
Service d’Endocrinologie
Avenue G. Thérasse 1
B-5530 Yvoir
081/42.32.85
corinne.jonas@uclouvain.be
Références
1. Lacroix A. ACTH-independent macronodular adrenal hyperplasia. Best Pract Res Clin Endocrinol Metab 2009 ;23:245-59.
ouvrir dans Pubmed
2. El Ghorayeb N, Bourdeau I, Lacroix A. Multiple aberrant hormone receptors in Cushing's syndrome. Eur J Endocrinol 2015; 173:M45-60.
ouvrir dans Pubmed
3. Chiodini I. Clinical review: Diagnosis and treatment of subclinical hypercortisolism. J Clin Endocrinol Metab 2011; 96:1223-36.
ouvrir dans Pubmed
4. Drougat L, Espiard S, Bertherat J. Genetics of primary bilateral macronodular adrenal hyperplasia: a model for early diagnosis of Cushing's syndrome? Eur J Endocrinol 2015; 173:M121-31.
ouvrir dans Pubmed
5. Vassilatou E, Vryonidou A, Ioannidis D, Paschou SA, Panagou M, Tzavara I. Bilateral adrenal incidentalomas differ from unilateral adrenal incidentalomas in subclinical cortisol hypersecretion but not in potential clinical implications. Eur J Endocrinol 2014 ; 171:37-45.
ouvrir dans Pubmed
6. Libé R, Coste J, Guignat L, Tissier F, Lefebvre H, Barrande G et al. Aberrant cortisol regulations in bilateral macronodular adrenal hyperplasia: a frequent finding in a prospective study of 32 patients with overt or subclinical Cushing's syndrome. Eur J Endocrinol 2010;163:129-38.
ouvrir dans Pubmed
7. Hofland J, Hofland LJ, van Koetsveld PM, Steenbergen J, de Herder WW, van Eijck CH et al. ACTH-independent macronodular adrenocortical hyperplasia reveals prevalent aberrant in vivo and in vitro responses to hormonal stimuli and coupling of arginine-vasopressin type 1a receptor to 11β-hydroxylase. Orphanet J Rare Dis 2013; 13; 8:142.
ouvrir dans Pubmed
8. Mircescu H, Jilwan J, N'Diaye N, Bourdeau I, Tremblay J, Hamet P et al. Are ectopic or abnormal membrane hormone receptors frequently present in adrenal Cushing's syndrome? J Clin Endocrinol Metab 2000; 85:3531-6.
ouvrir dans Pubmed
9. Louiset E, Duparc C, Young J, Renouf S, Tetsi Nomigni M, Boutelet I et al. Intraadrenal corticotropin in bilateral macronodular adrenal hyperplasia. N Engl J Med 2013; 369:2115-25.
ouvrir dans Pubmed
10. Miyamura N, Taguchi T, Murata Y, Taketa K, Iwashita S, Matsumoto K et al. Inherited adrenocorticotropin-independent macronodular adrenal hyperplasia with abnormal cortisol secretion by vasopressin and catecholamines: detection of the aberrant hormone receptors on adrenal gland. Endocrine 2002 ;19:319-26.
ouvrir dans Pubmed
11. Dekkers OM, Horváth-Puhó E, Jørgensen JO, Cannegieter SC, Ehrenstein V, Vandenbroucke JP et al. Multisystem morbidity and mortality in Cushing's syndrome: a cohort study. J Clin Endocrinol Metab 2013; 98:2277-84.
ouvrir dans Pubmed
12. Wagenmakers MA, Netea-Maier RT, Prins JB, Dekkers T, den Heijer M, Hermus AR. Impaired quality of life in patients in long-term remission of Cushing's syndrome of both adrenal and pituitary origin: a remaining effect of long-standing hypercortisolism? Eur J Endocrinol 2012;167:687-95.
ouvrir dans Pubmed
13. Di Dalmazi G, Pasquali R, Beuschlein F, Reincke M. Subclinical hypercortisolism: a state, a syndrome, or a disease? Eur J Endocrinol 2015; 173:M61-71.
ouvrir dans Pubmed
14. Tauchmanovà L, Rossi R, Biondi B, Pulcrano M, Nuzzo V, Palmieri EA et al. Patients with subclinical Cushing's syndrome due to adrenal adenoma have increased cardiovascular risk. J Clin Endocrinol Metab 2002 ; 87:4872-8.
ouvrir dans Pubmed
15. Di Dalmazi G, Vicennati V, Garelli S, Casadio E, Rinaldi E, Giampalma E et al. Cardiovascular events and mortality in patients with adrenal incidentalomas that are either non-secreting or associated with intermediate phenotype or subclinical Cushing's syndrome: a 15-year retrospective study. Lancet Diabetes Endocrinol 2014; 2:396-405.
ouvrir dans Pubmed
16. Debono M, Bradburn M, Bull M, Harrison B, Ross RJ, Newell-Price J. Cortisol as a marker for increased mortality in patients with incidental adrenocortical adenomas. J Clin Endocrinol Metab 2014; 99:4462-70.
ouvrir dans Pubmed
17. Debillon E, Velayoudom-Cephise FL, Salenave S, Caron P, Chaffanjon P, Wagner T et al. Unilateral Adrenalectomy as a First-Line Treatment of Cushing's Syndrome in Patients With Primary Bilateral Macronodular Adrenal Hyperplasia. J Clin Endocrinol Metab 2015; 100:4417-24.
ouvrir dans Pubmed
18. Preumont V, Mermejo LM, Damoiseaux P, Lacroix A, Maiter D. Transient efficacy of octreotide and pasireotide (SOM230) treatment in GIP-dependent Cushing's syndrome. Horm Metab Res 2011 ;43:287-91.
ouvrir dans Pubmed
19. Ritzel K, Beuschlein F, Mickisch A, Osswald A, Schneider HJ, Schopohl J et al. Outcome of bilateral adrenalectomy in Cushing's syndrome: a systematic review. J Clin Endocrinol Metab 2013; 98:3939-48.
ouvrir dans Pubmed
20. Assié G, Libé R, Espiard S, Rizk-Rabin M, Guimier A, Luscap W et al. ARMC5 mutations in macronodular adrenal hyperplasia with Cushing's syndrome. N Engl J Med 2013; 369:2105-14.
ouvrir dans Pubmed
21. Nieman LK, Biller BM, Findling JW, Newell-Price J, Savage MO, Stewart PM et al. The diagnosis of Cushing's syndrome: an Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab 2008; 93:1526-40.
ouvrir dans Pubmed