INTRODUCTION
L’Epstein-Barr virus, ou herpès virus humain 4, est un agent infectieux bien connu de la famille des herpès virus (1). Il a été découvert par M.A. Epstein, Y. Barr & B. Achong et fut le premier virus humain oncogène découvert. Environ 95% de la population mondiale en est porteur et la plupart l’a contracté durant l’enfance par transmission salivaire (2). L’infection est généralement asymptomatique mais si la primo-infection survient durant l’adolescence ou après, 25 à 50% des patients développeront le syndrome de mononucléose infectieuse (1). Le diagnostic repose sur l’examen clinique, avec la triade classique fièvre + pharyngite + adénopathies. On retrouve également la présence de lymphocytes réactionnels et d’anticorps hétérophiles dans les analyses sanguines. Toutefois, l’EBV peut être responsable d’une série de complications comme le lymphome de Burkitt ou le syndrome lympho-prolifératif lié à l’X, surtout chez des patients immunodéprimés. Ce virus peut mener à différentes complications neurologiques : encéphalite/encéphalomyélite, encéphalomyélite aigüe disséminée, myélite transverse, méningite, cérébellite, paralysie d’un nerf crânien, neuropathie périphérique, syndrome d’Alice au pays des merveilles1, Guillain-Barré et autres pathologies auto-immunes (3).
L’encéphalomyélite aigüe disséminée (ADEM) est l’une de ces complications post-EBV et est caractérisée par une démyélinisation pouvant affecter tout segment du système nerveux central, avec un taux de mortalité accru pour une lésion localisée au tronc cérébral (4). Bien que les processus menant à la pathologie soient encore inconnus, il s’agirait probablement d’une réponse auto-immune plutôt que d’une conséquence de la réplication virale (5). L’antigène nucléaire de l’EBV (EBNA) contient un pentapeptide très similaire à un morceau de protéine de myéline. Les anticorps et la réaction inflammatoire engendrée par le virus cibleraient alors aussi la myéline, causant l’ADEM. C’est une présentation rare de l’infection et la sévérité varie de cas banals au décès (6). Les patients se présentent généralement avec fièvre, confusion, altération de l’état de conscience et signes neurologiques multifocaux. Ces manifestations peuvent apparaître avant, voir même sans, les symptômes de la mononucléose ce qui rend le diagnostic difficile. L’incidence des encéphalites au sens large à EBV est d’environ 7% parmi les enfants hospitalisés et la mortalité de 0.1 à 1% (3).
CAS CLINIQUE
Une enfant de 25 mois est admise aux urgences pour convulsions, vomissements et altération de l’état de conscience depuis quelques jours. Depuis 4 semaines, elle a une infection des voies respiratoires supérieures avec fièvre persistante (autour des 39°C), rhinopharyngite et otite gauche. Les parents de l’enfant ont déjà consulté plusieurs hôpitaux en raison de la persistance de la fièvre et de l’altération de l’état général de leur fille. Elle a déjà été traitée par amoxicilline pour une angine, mais l’antibiotique a été stoppé 13 jours plus tôt lorsqu’une primo-infection par EBV est diagnostiquée sur base des sérologies. L’enfant est en ordre de vaccination et ses antécédents médicaux sans particularités. À l’admission, l’examen clinique révèle une pâleur cutanée, une angine avec hypertrophie amygdalienne sévère, mais pas d’adénopathies cervicales. Elle est somnolente et présente une instabilité posturale sans aucun autre signe neurologique. Son périmètre crânien de 50 cm se situe dans les limites supérieures de la normale (P90-97). Ses paramètres vitaux sont normaux pour l’âge hormis une fièvre à 39.2°C.
À la prise de sang, il n’y a pas de syndrome inflammatoire, mais une lymphocytose à 15.0 x 10³/mm³ (Nl : 4.0-10.0 x 10³/mm³), une élévation modérée des enzymes hépatiques et une anémie microcytaire par carence martiale. Les hémocultures et la culture d’urine sont revenues stériles, mais les Ig M & G pour l’antigène de la capside virale (VCA) de l’EBV sont positifs alors que les Ig G contre l’antigène nucléaire (EBNA) non. D’autres sérologies reviennent aussi négatives (cytomegalovirus, varicella zoster virus, herpes simplex virus 1 & 2, Chlamydia pneumoniae, oreillon, rougeole, ECHO virus). Elle a été initialement traitée par acyclovir et ceftriaxone jusqu’à l’obtention des résultats. Un CT-scanner sans contraste a révélé une hydrocéphalie sévère, probablement liée à un processus chronique vu l’évolution du périmètre crânien. L’EEG a démontré un ralentissement global du tracé, mais pas de signe de focalisation ou d’activité épileptiforme. Une IRM cérébrale réalisée en urgence a montré d’importantes lésions démyélinisantes autour des pédoncules cérébraux avec atteinte du tronc cérébral, lésions compatibles avec une méningo-encéphalite. L’enfant a rapidement été admise en salle d’opération pour y réaliser une dérivation ventriculaire externe, ce qui a mené à une amélioration clinique initiale puisqu’elle regagna la parole et la marche. Le liquide céphalo-rachidien obtenu lors de l’opération a été analysé et a révélé une pléiocytose de 46 cellules/mm³ (Nl : < 5/mm³) à prédominance lymphocytaire (92%), une protéinorachie de 0.98 g/L (Nl : 0.15-0.40 g/L) et une glycorachie normale. Les cultures et sérologies (EBV, HZV, HSV 1 & 2, Bartonella henselae, Mycoplasma pneumoniae et Mycobacterium tuberculosis) sur le LCR n’étaient pas contributives. Deux jours après la chirurgie, l’enfant devint bradycarde et aphasique. Elle présenta alors des convulsions de l’hémicorps gauche. L’examen clinique montra un myosis, une aréflexie avec raideur des membres gauches ainsi qu’un signe de Babinski positif bilatéralement. A l’arrivée des soins intensifs, elle était inconsciente et son score de Glasgow était de 5/15 (Y 1, V 1, M 3). Elle a été intubée et transférée aux soins intensifs où elle demeura dans un coma durant 3 semaines. De fortes doses de méthylprednisolone IV furent administrées, suivies de plasmaphérèses en raison du manque de réponse au traitement initial. Elle développa de l’hypertension artérielle et fit un arrêt cardiaque. L’IRM de contrôle, réalisée 17 jours après la première, montra une importante progression des lésions. L’extension concernait les noyaux gris centraux, le tronc cérébral, le pont et les pédoncules cérébelleux. On retrouvait également des lésions dans la moelle allongée, une dilatation ventriculaire majeure ainsi que des signes de souffrance péri et rétro-ventriculaire associés à une importante collection sous-durale gauche. A ce stade, l’examen clinique révéla une anisocorie. Sachant la gravité de sa situation et vu l’échec du traitement curatif, une prise en charge palliative fut instaurée. L’enfant a été extubée et la sédation majorée, ce qui mena rapidement à une insuffisance respiratoire et une mort paisible. La biopsie cérébrale montra un processus cérébral démyélinisant et inflammatoire, compatible avec une encéphalomyélite aigüe disséminée.
DISCUSSION
L’ADEM est un processus inflammatoire et démyélinisant du système nerveux central survenant essentiellement chez des enfants et qui évolue sur un mode monophasique. Toutefois, une minorité des patients présentent un mode multiphasique/ avec rechutes. Il existe alors 2 épisodes d’ADEM séparés de minimum 3 mois et aucun autre événement. Cette 2e rechute peut consister en la réapparition ou en la nouvelle apparition de déficits neurologiques ou de lésions à l’imagerie. Dans la forme classique, la maladie débute par la survenue aigüe ou subaigüe de symptômes neurologiques multifocaux et d’une encéphalopathie souvent précédés d’une infection ou d’une vaccination. Ici, l’enfant a contracté une infection à EBV et a par la suite présenté un épisode de convulsions associé à des signes d’encéphalopathie. La sévérité des manifestations cliniques peut progresser durant les trois premiers mois (7).
C’est une maladie rare dont 3 à 6 cas sont observés par an dans les centres médicaux du Royaume-Uni, des USA et d’Australie (8). Il ne semble pas exister de sensibilité ethnique particulière, mais les études tendent à montrer une légère prédominance masculine. L’âge moyen de survenue se situe entre 6 et 8 ans, la pathologie étant prouvée moins commune en dessous de 2 ans (5). Notre cas est donc atypique puisqu’il concerne une fillette de 25 mois.
L’origine de l’ADEM rapporté était une primo infection par EBV. La majorité des cas est en effet précédée par une infection virale, plus rarement bactérienne, affectant typiquement les voies aériennes supérieures. Cela suit également une distribution saisonnière avec la plupart des cas survenant à l’hiver et au printemps, pour le nôtre le mois de Janvier (8). Plusieurs pathogènes ont été identifiés parmi lesquels : EBV, CMV, HSV, VZV, HIV, Beta hemolytic Streptococcus, Mycoplasma pneumoniae, Chlamydia pneumoniae, Borrelia burgdorferi, … (9).
Par ailleurs, moins de 5% des ADEM surviennent à la suite d’une vaccination, la plus fréquente étant associée au vaccin rougeole-rubéole-oreillon. Il est pourtant crucial de préciser que l’incidence d’ADEM à la suite d’une rougeole (1 cas pour 1000 infectés) est beaucoup plus élevée que celle associée à la vaccination RRO (2 cas pour 1 million de vaccinés) (6).
Bien que la pathogenèse ne soit pas entièrement élucidée, l’ADEM semble résulter d’un processus auto-immun déclenché par un stimulus environnemental qui mène, chez des individus prédisposés génétiquement, à la destruction de myéline. L’hypothèse principale serait qu’un certain antigène de la myéline (proteolipid protein, myelin oligodendrocyte protein ou myelin basic protein) partage des propriétés antigéniques similaires à celles du pathogène. Dans notre cas, l’EBV est un parfait exemple de cette cross-réaction. L’antigène nucléaire de l’EBV (EBNA) contient un pentapeptide très similaire à un épitope de la myelin basic protein. Les anticorps dirigés contre l’EBV ainsi que la réaction immunitaire cellulaire au virus se trompent de cible et attaquent cette protéine de myéline, causant l’ADEM. Ce phénomène connu sous le nom de « mimétisme moléculaire » n’implique pas toujours une réplication virale active. Les études tendent à montrer que les enfants atteints d’ADEM ont des lignées de lymphocytes T réagissant 10 fois plus à la myelin basic protein que ceux qui souffrent de simple encéphalite. D’autres hypothèses ont été proposées comme l’insertion de polypeptides par le virus dans la membrane cellulaire de l’hôte, ce qui déclencherait une réaction auto-immune, ou encore la capacité du virus à relâcher des antigènes de myéline en circulation. La compréhension de cette pathologie est toujours l’objet de recherches scientifiques (2-5-6).
Notre patiente avait une fièvre persistante depuis 4 semaines avant son premier épisode de convulsion. Ceci est observé dans 50 à 75 % des cas. Parmi les autres symptômes, on retrouve l’association de fièvre, céphalée, vomissements et méningisme, que l’on retrouve souvent chez les enfants hospitalisés pour ADEM (6). Toutefois, ces symptômes sont aspécifiques ce qui rend impossible la prédiction de survenue d’une ADEM. Dans notre cas, ces signes pouvaient simplement être attribués à l’infection par EBV diagnostiquée 2 semaines avant la survenue des symptômes neurologiques. La présentation classique de l’ADEM associe une encéphalopathie rapidement évolutive à des déficits neurologiques multifocaux (9). Notre patiente était malade depuis un certain temps, mais dès lors qu’elle a développé sa convulsion, son état général s’est rapidement dégradé ce qui est également un élément clé de l’ADEM. Les signes neurologiques atteignent généralement leur sévérité maximale autour du 4e ou 7e jour suivant le premier signe neurologique (6). Bien que notre cas présentait toutes les manifestations communes d’encéphalopathie (irritabilité, confusion et évolution de l’état de conscience de somnolence et léthargie à un coma franc), elle a également développé des convulsions ce qui est moins fréquent. Seulement 1/3 des patients atteints d’ADEM en font (6-10). En plus de son encéphalopathie, la petite fille avait un tableau d’atteinte motrice incluant spasticité, hyperréflexie, signe de Babinski positif bilatéralement, incapacité à réaliser les mouvements fins et mouvements anormaux. Tous ces signes indiquent une atteinte pyramidale. Ceci est aussi une manifestation très fréquente de l’ADEM, tout comme l’ataxie cérebelleuse, les neuropathies crâniennes ou encore l’hémiparésie. Par ailleurs, notre patiente avait également des manifestations atypiques : elle était aphasique et avait des attitudes anormales comme une hyperextension du bras gauche. La phase aigüe d’une ADEM dure généralement 2 à 4 semaines avec une possible détérioration neurologique initiale, mais la plupart guériront complètement.
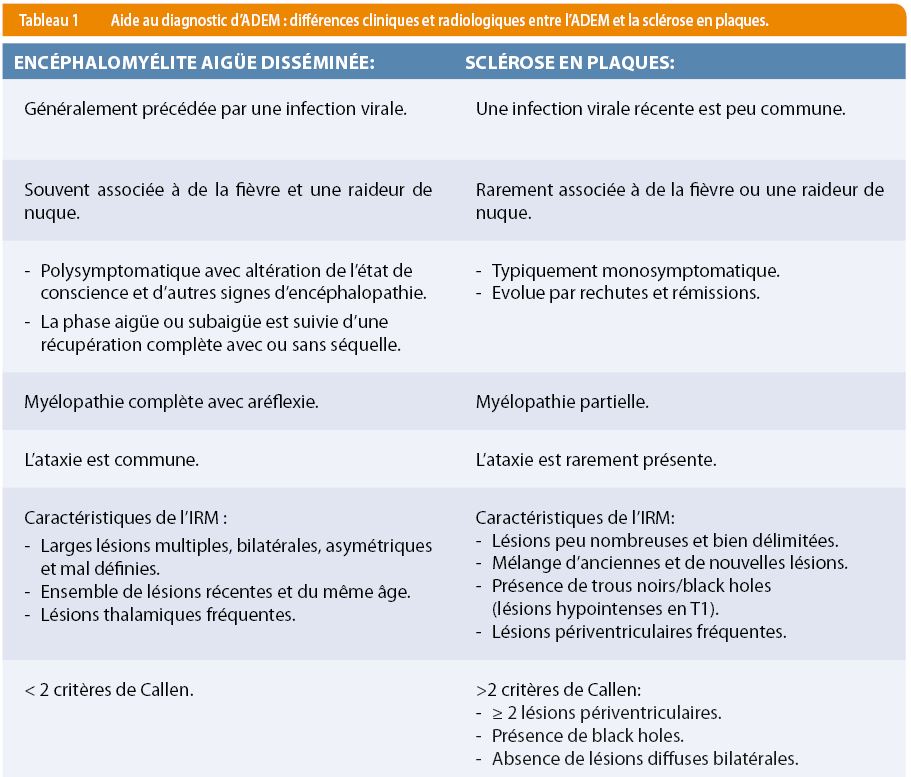
ll existe plusieurs autres pathologies centrales démyélinisantes chez l’enfant très similaires à l’ADEM de par leur présentation clinique et leur origine auto-immune post-infectieuse. Parmi celles-ci, la plus proche de l’ADEM est la sclérose en plaques, mais on note également la myélite transverse, la névrite optique et la neuromyélite optique (ou maladie de Devic). Pour rendre le diagnostic plus facile, la société pédiatrique internationale de sclérose en plaques a réalisé une large étude afin d’élaborer des critères diagnostics. Ainsi, l’ADEM requiert un premier évènement clinique associant des symptômes d’encéphalopathie non-expliqués par la fièvre, une maladie systémique ou un état post-critique épileptique, à des signes neurologiques multifocaux. Le diagnostic se base également sur les signes typiques visualisés à l’IRM cérébrale ainsi que sur l’absence de nouvelles lésions à l’IRM de contrôle à réaliser 3 mois plus tard. Le fait que l’encéphalopathie survienne au même moment que la phase aigüe aide à différencier l’ADEM de la sclérose en plaques (6-7). Certaines différences peuvent également aider (11) (Tableau 1).
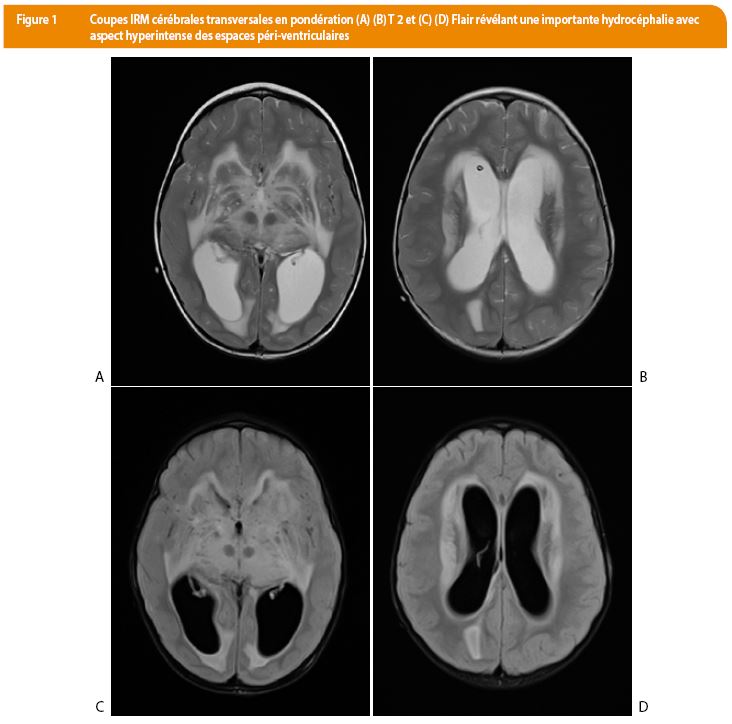
Le diagnostic d’ADEM est donc essentiellement clinique et repose sur les critères diagnostics précédemment décrits. Les CT-scanners ne sont pas utiles puisque les lésions peuvent ne pas y être révélées. Il a cependant été réalisé dans notre cas au vu du périmètre crânien à la limite supérieure et a révélé une importante hydrocéphalie. L’hydrocéphalie n’étant pas une caractéristique de l’ADEM et au vu du périmètre crânien en augmentation, il est probable qu’elle était ici liée à une affection chronique indépendante dont l’étiologie reste indéterminée (Figure 1).
L’IRM est l’imagerie de choix pour visualiser une ADEM et détecte les lésions dans les 72h suivant le premier symptôme neurologique. On préférera les séquences T2 et FLAIR ou encore l’utilisation de contraste au gadolinium. Comme ce fut ici le cas, on visualise de larges lésions diffuses, asymétriques et mal définies de la substance blanche, souvent associées à de l’œdème. Chez les enfants, on retrouve aussi souvent des lésions de la substance grise et il n’est pas rare de trouver une atteinte du thalamus, des noyaux gris centraux, du tronc cérébral ou de la moelle épinière (1-5-6-11).
Les investigations devraient aussi inclure:
- une prise de sang de routine qui montre généralement un syndrome inflammatoire et une hyperleucocytose à prédominance lymphocytaire, ce qui était le cas chez notre enfant ;
- la recherche de divers agents pathogènes sur frottis de gorge et du nasopharynx, coproculture, analyse de LCR et sérologies sanguines. Ceci n’était pas contributif chez notre patiente ;
- une ponction lombaire, ce qui ne fut pas réalisé ici en raison de la dérivation ventriculaire qui nous permit d’analyser le LCR. Dans la majorité des cas, on retrouvera une pléiocytose et/ou une majoration de la protéinorachie et des marqueurs inflammatoires. Parfois, on aura même une analyse strictement normale du LCR (1-5-6-8) ;
- l’électroencéphalogramme n’est pas un outil diagnostique, mais peut montrer un ralentissement du tracé corrélé à l’encéphalopathie, comme ce fut le cas pour notre patiente (6).
Pour ce qui est de la prise en charge, les enfants doivent initialement être traités par acyclovir et antibiotiques à large spectre jusqu’à ce qu’une infection par bactérie ou herpès soit exclue. Notre patiente a ainsi reçu de la ceftriaxone et de l’acyclovir. À cause du manque d’études cliniques, il n’existe actuellement pas de thérapie standard pour l’ADEM, mais il semble être recommandé d’administrer de fortes doses de glucocorticoïdes intraveineux aussi tôt que possible, même sous traitement par antibiotiques et acyclovir. Un enfant devrait recevoir soit de la méthylprednisolone IV (10 à 30 mg/kg/j avec un maximum de 1000mg/j), soit de la dexaméthasone IV (1mg/kg/j) durant 5 jours. La méthylprednisolone a démontré une meilleure efficacité dans certaines études et notre enfant a donc été traitée avec ce médicament. Un relai par glucocorticoïdes per os peut ensuite être poursuivi pendant 4 à 6 semaines chez les enfants encore symptomatiques. On démarre avec une dose de 1mg/kg/j (maximum 60mg/j) de prednisone que l’on réduit ensuite de 10mg/5 jours durant les 4 à 6 semaines. Certains recommandent l’utilisation des immunoglobulines IV comme traitement de seconde ligne chez les enfants n’ayant pas répondu aux corticoïdes. D’après les études, un total de 2g/kg réparti sur 3 jours devrait être administré. Une autre option thérapeutique est la plasmaphérèse. Elle est indiquée chez les enfants présentant une myélite transverse étendue et pour laquelle le traitement par corticoïdes a échoué ou encore comme troisième ligne de traitement après les Ig IV. Un total de 6 plasmaphérèses avec 1 à 1.5 de volume plasmatique échangé est recommandé. Au vu de la sévérité et de la progression rapide des lésions chez notre patiente, des plasmaphérèses furent initiées après l’échec du traitement par glucocorticoïdes, mais ne menèrent à aucune amélioration (1-5-12).
La plupart des cas pédiatriques d’ADEM ont un excellent pronostic avec une récupération lente, mais complète. Dans plusieurs études, les ADEM traitées par glucocorticoïdes IV sont associées à une guérison complète sans déficit neurologique dans 60 à 90 % des cas. L’IRM de contrôle montre généralement une disparition des lésions, mais on retrouve parfois des plages de démyélinisation et de gliose. Les patients ne récupérant pas complètement présentent le plus souvent des complications mineures comme des céphalées, troubles comportementaux, baisse de l’acuité visuelle ou encore une hémiparésie modérée. Certains développent des séquelles plus sévères incluant épilepsie partielle, handicap mental, paraparésie sévère ou troubles moteurs. D’autres encore évoluent plus rarement vers un ADEM multiphasique ou vers une sclérose en plaques. Les décès restent l’exception et surviennent généralement par œdème ou hémorragies cérébrales. Bien que la mortalité des ADEM post-infectieuses dans la population pédiatrique ne dépasse les 5%, notre petite patiente faisait malheureusement partie de ce groupe (5-12).
CONCLUSION
Bien qu’une ADEM a un bon pronostic chez l’enfant, c’est une affection rare pouvant mener à de sévères conséquences, voire même au décès. Par conséquent, elle doit être évoquée chez tout enfant qui, à la suite d’une infection ou d’une vaccination, présente de la fièvre, une encéphalite aigue associée à des signes neurologiques et dont les analyses de sang ou de LCR révèlent de l’inflammation. Une IRM cérébrale doit être réalisée. La prise en charge initiale doit inclure l’utilisation d’acyclovir et d’antibiotiques à large spectre jusqu’à l’exclusion d’une infection bactérienne ou herpétique tout en administrant de fortes doses de glucocorticoïdes IV. Pour ceux qui ne répondraient pas suffisamment au traitement, les immunoglobulines IV devraient être considérées. Les plasmaphérèses sont à réserver comme traitement de 3e ligne ou en cas de myélite transverse expansive. Comme il n’existe aucun marqueur biologique ou clinique et qu’elle présente beaucoup de similarités avec la sclérose en plaques, le diagnostic d’ADEM reste difficile et un suivi à long-terme doit être programmé. La survenue de rechutes nous orientera alors vers un autre diagnostic comme l’ADEM multiphasique ou la sclérose en plaques.
AFFILIATIONS
1 Faculté de médecine et médecine dentaire, Université catholique de Louvain, 50, Avenue E. Mounier, 1200 Bruxelles.
2 Service de soins intensifs pédiatriques, Cliniques Universitaires Saint-Luc, 10, Avenue Hippocrate, 1200 Bruxelles.
3 Service de pédiatrie, Grand Hôpital de Charleroi : Notre-Dame, 3, Grand Rue, 6000 Charleroi.
CORRESPONDANCE
Dr. MARIE-LAURE OBERWEIS
Faculté de médecine et médecine dentaire, Université catholique de Louvain, 50, Avenue E. Mounier, 1200 Bruxelles.
RÉFÉRENCES
- Zhang S, Feng J, Shi Y. Transient widespread cortical and splenial lesions in acute encephalitis/encephalopathy associated with primary Epstein-Barr virus infection. Int J Infect Dis. 2016 Jan;42:7-10. doi: 10.1016/j.ijid.2015.11.009. Ouvrir dans PubMed
- Holck Draborg A, Duus K, Houen,G. Epstein-Barr Virus in systemic autoimmune diseases: review article. J Immunol Res (Hindawi Publishing Corporation – Clinical and Developmental Immunology), Volume 2013, Article ID 535738. http://dx.doi.org/10.1155/2013/535738 Ouvrir dans PubMed
- Bolis V, Karadedos C, Chiotis I, Chaliasos N, Tsabouri S. Atypical manifestations of Epstein-Barr Virus in children: a diagnostic challenge. J Pediatr (Rio J). 2016 Mar-Apr;92(2):113-21. doi: 10.1016/j.jped.2015.06.007. Ouvrir dans PubMed
- Abul-Kasim K, Palm L, Maly P, Sundgren, PC. The neuroanatomic localization of Epstein-Barr virus encephalitis may be a predictive factor for its clinical outcome: a case report and review of 100 cases in 28 reports. J Child Neurol. 2009 Jun;24(6):720-6. doi: 10.1177/0883073808327842. Ouvrir dans PubMed
- Mohsen H, Abu Zeinah GF, Elsotouhy AH, Mohamed K. Acute disseminated encephalomyelitis following infectious mononucleosis in a toddler: case report. BMJ. Doi:10.1136/bcr-2013-010048. Ouvrir dans PubMed
- Lotze TE, Chadwick DJ. Acute disseminated encephalomyelitis in children: pathogenesis, clinical features and diagnosis. In: UpToDate, Patterson, MC., UpToDate, Dashe, JF.
- Krupp LB, Tardieu M, Amato, MP et al. International Pediatric Multiple Sclerosis Study Group criteria for pediatric multiple sclerosis and immune-mediated central nervous system demyelinating disorders: revisions to the 2007 definitions. Multiple Sclerosis. 2013, 19: 1261. Ouvrir dans PubMed
- Murthy SN, Faden HS, Cohen ME, Bakshi R. Acute disseminated encephalomyelitis in children. Pediatrics. 2002, 110: e21. Ouvrir dans PubMed
- Tenembaum S, Chitnis T, Ness J, Hanh JS. et al. Acute disseminated encephalomyelitis. Neurology. 2007, 68: s23. Ouvrir dans PubMed
- Davis LE, Booss J. Acute disseminated encephalomyelitis in children: a changing picture. Pediatr Infect Dis J. 2003 Sep;22(9):829-31. Ouvrir dans PubMed
- Lotze TE. Differential diagnosis of acute central nervous system demyelination in children. In: UpToDate, Patterson, MC., UpToDate, Dashe, JF. (Accessed on May, 05, 2017).
- Lotze TE, Chadwick DJ. Acute disseminated encephalomyelitis in children: treatment and prognosis. In: UpToDate, Patterson, MC., UpToDate, Dashe, JF. (Accessed on May, 05, 2017).