Introduction
L’hépatite fulminante ou insuffisance hépatique aiguë est une maladie rare et complexe qui se manifeste par une coagulopathie (rapport international normalisé, INR ≥ 1,5) et une altération de l’état mental (encéphalopathie hépatique) due à un déclin rapide de la fonction hépatique chez un patient sans maladie hépatique préexistante. Le début et la durée de la maladie doivent être inférieurs à 26 semaines (1). Les causes en sont multiples : les virus hépatotropes (principalement les virus des hépatites A et B), les toxiques et les médicaments en étant les plus fréquentes (1). La prise en charge de l’hépatite fulminante passe par une recherche rigoureuse de l’étiologie afin d’initier un éventuel traitement spécifique précocement pour améliorer les chances de récupération de la fonction hépatique et la survie. Néanmoins, certaines étiologies sont plus rares, de diagnostic difficile, avec des conséquences néfastes en termes de pronostic. L’hépatite herpétique en est un exemple avec un taux de mortalité majeur dû à l’insuffisance hépato-cellulaire aigue et à ses complications telles que l’insuffisance rénale aigue, les coagulopathies à l’origine d’hémorragies et le syndrome hémophagocytaire réactionnel (2).
Vignette clinique
Un homme de 36 ans se présente aux Urgences pour altération brutale de l’état général avec fièvre à 39°C, vomissements et myalgies depuis 2 jours.
Le patient n’a pas d’antécédents particuliers, ne prend pas de traitement chronique et est allergique aux pénicillines.
L’interrogatoire révèle une infection ORL chez les deux enfants du patient, une semaine avant le début des symptômes. À noter l’absence de voyage récent, de rapports sexuels non protégés ou de consommation d’aliments inhabituels. Un traitement par paracétamol à la dose de 4 grammes par jour ainsi que de l’ibuprofène à la dose de 400 mg trois fois par jour ont été débutés il y a 48 heures pour ce syndrome pseudo-grippal.
Les paramètres d’admission sont les suivants : pression artérielle à 130/80 mmHg, température à 38.6°C, fréquence cardiaque à 110 battements par minute et saturation en oxygène à l’air ambiant à 92 %. L’examen clinique est banal hormis une sensibilité épigastrique à la palpation abdominale. La biologie initiale montre une hépatite cytolytique avec des GOT à 916 U/L (valeur normale (VN) : <50 U/L) et des GPT à 864 U/L (VN : <50 U/L), une élévation des LDH à 1865 U/L (VN :135-225 U/L), un syndrome inflammatoire avec une CRP à 299 mg/L (VN : <5.0 mg/L), une lymphopénie à 340 /µL (VN : 1100-4500 /µL) et une thrombopénie à 147000 /µL (VN : 150000-370000 /µL). La paracétamolémie est de 6.1 µg/ml (VN : < 10 µg/ml) et l’INR est à 1.13 (VN : < 1.30). Le reste de la biologie et l’abdomen à blanc sont sans particularités.
Le patient est admis dans le service de maladies infectieuses pour poursuivre la prise en charge. Un traitement par ciprofloxacine est initié empiriquement pendant 48 heures dans l’hypothèse d’une infection bactérienne d’origine digestive au sens large. L’évolution clinique est marquée par une dégradation clinique rapide avec la persistance d’épisodes fébriles à 40°C nécessitant l’administration récurrente de paracétamol ; ainsi qu’une détérioration biologique avec une aggravation de la cytolyse (GOT à 5993 U/L et GPT à 2986 U/L). En outre, apparaissent une neutropénie à 450 /µL (VN : 1500 - 7700 /µL), une hypoalbuminémie à 30 g/L (VN : 35-52 g/L), une diminution du facteur V à 46% (VN : 62-150%) et une perturbation de l’hémostase avec un INR à 1.65 (VN : < 1.50). La lymphopénie et la thrombopénie s’aggravent. La ferritinémie est alors mesurée à 42300 µg/L (VN : 30-400 µg/L), le fibrinogène à 2.11 g/L (VN : 2.00-4.00 g/L) et les triglycérides à 106 mg/dL (VN : <150 mg/dL). Dans ce contexte, une ponction biopsie de moelle est réalisée dans l’hypothèse d’un syndrome hémophagocytaire au vu de l’hyperferritinémie, de la bi-cytopénie (neutropénie et thrombopénie) et de la fièvre persistante. Cette dernière mettra en évidence une moelle d’aspect essentiellement réactionnel avec une activité macrophagique augmentée sans signes d’hémophagocytose. Diverses sérologies (hépatites A, B, C et E, virus d’Epstein-Barr (EBV), cytomégalovirus (CMV), virus d’immunodéficience humaine (VIH), HSV1, Parvovirus B19, Mycoplasma pneumoniae, Legionella pneumophila, Chlamydia pneumoniae, Coxiella burnetti, Bartonella henselae, Brucella, Leptospira et Toxoplasma gondii) et PCR sanguines (EBV et CMV) sont réalisées sans apporter de confirmation diagnostique. L’ensemble des hémocultures réalisées sont revenues négatives. Une échographie hépatique est réalisée ne montrant pas de signes d’hépatopathie chronique ni d’anomalies vasculaires hépatiques ni de splénomégalie. L’arrêt du paracétamol et un traitement par acétylcystéine s’avèrent inefficaces dans l’hypothèse d’une intoxication au paracétamol malgré une paracétamolémie inférieure à 5 µg/ml.
Un traitement empirique par aciclovir est alors initié et un transfert dans un centre de greffe hépatique organisé. Lors du séjour aux soins intensifs, le patient développe progressivement des mouvements stéréotypés et du jargonisme justifiant une résonance cérébrale ne montrant pas de signes d’encéphalopathie et une ponction lombaire sur laquelle la PCR HSV1 s’avère positive de même que la PCR sanguine. Un syndrome hémophagocytaire est, à nouveau, suspecté au vu de l’aggravation des cytopénies, l’hyperferritinémie persistante à > 10.000 µg/L, une hypertriglycéridémie à 538 mg/dL et une hypofibrinogénémie à 80 mg/dL (VN : 150-450 mg/dL). Une nouvelle ponction biopsie médullaire confirmera cette fois-ci la présence d’une hémophagocytose et par conséquent, la présence d’un syndrome hémophagocytaire. La biopsie hépatique démontrera ultérieurement la présence d’une nécrose sub-massive du parenchyme hépatique et la PCR HSV1 s’y avérera également positive. Le séjour aux soins intensifs durera 12 jours au cours duquel le patient présentera plusieurs complications : une insuffisance rénale aigue avec un pic de créatinine à 3.11 mg/dL (VN : 0.60-1.30 mg/dL) et une bactériémie à S.aureus oxacilline sensible secondaire à une pneumonie basale droite apparue 9 jours après le début de l’infection herpétique. Le traitement par aciclovir a été poursuivi aux soins intensifs puis en unité de maladies infectieuses en association avec une antibiothérapie par céfazoline pour une durée totale de 18 jours. Une amélioration des paramètres hépatiques fut rapidement constatée ainsi qu’une normalisation de l’INR (1.12 (VN : 0.80-1.20)) et du taux de facteur V (>140 % (VN : 70-140 %)). Par conséquent, l’inscription sur la liste de transplantation hépatique ne fut pas nécessaire.
Discussion
HSV1 est un virus à ADN double brin de la sous famille des Herpesviridae. Il est responsable, dans la majorité des cas, d’infections oro-labiales après un contact avec des muqueuses buccales infectées et, dans une moindre mesure, d’infections au niveau de la sphère génitale suite à un contact oro-génital. La présentation clinique la plus classique consiste en l’apparition de vésicules ou d’ulcérations douloureuses. Dans la majorité des cas, la primo-infection survient dans l’enfance et est souvent asymptomatique. Par la suite, on peut observer des récidives périodiques sous forme d’infection plus localisée au niveau labial communément appelée « bouton de fièvre » (3). D’un point de vue épidémiologique, il s’agit d’une infection très fréquente. En 2016, James et al ont estimé à 3583.5 millions le nombre d’individus de la population mondiale âgés de 0-49 ans infectés par voie orale par l’HSV 1, soit une prévalence de 63.6 % (4).
Dans certaines circonstances, l’HSV1 peut provoquer des infections plus sévères dont l’exemple le plus couramment rencontré est l’encéphalite herpétique. Celle-ci représente 10 à 20 % des 20 000 patients atteints d’encéphalite virale chaque année aux États-Unis, avec une mortalité estimée entre 20 et 30 % et ce, même en cas de diagnostic et de traitement précoces (5). En France, l’HSV1 est la première cause d’encéphalite virale soit 42 % des encéphalites virales documentées dans une étude prospective de 253 patients (6). Comme nous le rapportons, l’HSV1 peut également être responsable d’une atteinte hépatique sévère à l’origine d’une insuffisance hépatique aigue, représentant 2 % des insuffisances hépatiques aigues associées à un virus (2). À titre comparatif, le virus de l’hépatite B en Europe y compte jusque 19 % (7).
L’hépatite herpétique survient plus fréquemment chez les patients immunodéprimés. Toutefois, les patients immunocompétents ne sont pas épargnés. Dans une étude rétrospective, 53 % des 137 cas rapportés étaient immunodéprimés, 24 % immunocompétents et 23% étaient des femmes enceintes (8).
Quatre théories ont été émises quant à la physiopathologie de cette atteinte. La première étant l’exposition à un inoculum viral important lors de la primo-infection dépassant le système immunitaire de l’hôte. La deuxième suggére une dissémination due à la défaillance des monocytes et des macrophages. La troisième évoque la possibilité d’une réinfection par une nouvelle souche virale réactivant une souche latente hépato-virulente. Enfin, la dernière évoque la possibilité d’une infection par un HSV1 hépato-virulent (2,9).
Les manifestations de l’hépatite herpétique sont aspécifiques en début d’infection, rendant le diagnostic difficile. En plus de la majoration des transaminases sans cholestase (hépatite anictérique), on peut retrouver une triade initiale comprenant de la fièvre, une leucopénie et une thrombopénie, mais également un rash herpétique dans 50 % des cas, une coagulopathie, une encéphalopathie ou encore une insuffisance rénale aigue (2,9). Ces manifestations ont été retrouvées successivement chez notre patient, à l’exception du rash.
Le diagnostic de l’hépatite herpétique se base sur la sérologie et la détection de l’ADN viral par polymerase chain reaction (PCR), la biopsie hépatique restant toutefois le gold standard pour confirmer l’atteinte hépatique malgré les difficultés potentielles de réalisation (coagulopathie, thrombopénie) (9). Les trois techniques diagnostiques ont été utilisées dans notre cas : la sérologie herpétique s’est positivée en cours d’hospitalisation confirmant la primo-infection et la biopsie a montré la présence d’une hépatite sub-massive nécrosante avec 60 % d’hépatocytes résiduels viables, avec une PCR HSV1 sanguine et hépatique positive.
L’hépatite herpétique est une pathologie avec un taux de mortalité élevé allant de 80 % à 90 % (2,9,10). Selon les études, ce dernier reste élevé (50 % de taux de mortalité) même après initiation du traitement (10). Il est donc important d’initier un traitement le plus rapidement possible en cas de suspicion clinique voire même de façon empirique face à une hépatite sévère afin de maximaliser les chances de survie et d’éviter la transplantation hépatique en urgence (9,11).
Hemophagocytic lymphohistiocytosis (HLH) ou syndrome hémophagocytaire (SH) est une entité clinico-pathologique mortelle caractérisée par une absence ou une diminution de la fonction des cellules Natural Killer (NK) et des lymphocytes T cytotoxiques à l’origine d’une activation immunitaire incontrôlée et inefficace. Cette activation conduit à une dysfonction multi-organique ainsi qu’à une prolifération et une activation des macrophages avec hémophagocytose dans tout le système réticulo-endothélial, à l’origine d’une pancytopénie, d’une hépatosplénomégalie et d’une lymphadénopathie (12). Il existe deux catégories de SH : le SH primaire (SHP), d’origine génétique, survenant durant l’enfance et le SH secondaire ou réactionnel (SHR), d’origine non génétique, survenant à tout âge (12-14). Ces deux catégories peuvent néanmoins s’entre-mêler : le SH primaire pouvant être déclenchées par des infections (ou tout autre événement activant l’immunité) et des mutations génétiques pouvant être mises en évidence dans le SH secondaire (14,15). Le terme « syndrome d’activation macrophagique » se définit comme un syndrome hémophagocytaire secondaire dont l’étiologie est rhumatologique (ex. : en cas de lupus érythémateux disséminé, de maladie de Sjögren, d’arthrite juvénile systémique, …) (13,14).
Les causes du SH secondaire sont nombreuses : les infections (50 %), les maladies auto-immunes (13 %) et les néoplasies toute origine confondue (47%) (14). Parmi les agents infectieux les plus rencontrés, les virus se trouvent en bonne place, en particulier les virus de la famille Herpes dont le chef de fil est l’EBV (66-69% des cas). Viennent ensuite le VIH, l’Influenza, le Parvovirus B19 et bien d’autres. A l’heure actuelle, HSV1 est une cause rare de SH secondaire, le nombre de cas s’y rapportant étant faible dans la littérature (12,15).
Anciennement, le diagnostic de SH était basé sur les critères diagnostiques de l’Histiocyte Society datant de 2004 qui étaient fondés sur des données pédiatriques, le SHP étant rencontré majoritairement chez les enfants (14). Le diagnostic était retenu si présence d’une mutation génétique évocatrice ou si présence de 5 critères sur 8 repris dans le tableau 1. Bien qu’ils présentent une importante spécificité (96%), ces critères sont peu sensibles (56.6%) et ne tiennent pas compte des manifestations cliniques ou biologiques propres à l’adulte. En outre, le déficit d’activité des cellules NK est surtout retrouvé dans les SHP (14). De ce fait, un diagnostic de SH est parfois retenu et un traitement initié même si les critères ne sont pas remplis.

En 2014, Fardet et al publient un score permettant de calculer le risque individuel d’avoir un SHR. Ce score appelé H-score est basé sur des données provenant d’une étude rétrospective multicentrique portant sur 312 patients. Il comprend 9 variables (3 cliniques, 5 biologiques et 1 cytologique) selon lesquelles est attribué un nombre de points (tableau 2). L’addition de ces points correspond à une probabilité de SHR variant de <1 % pour les scores ≤ 90 à > 99% pour les scores au-delà de 250 avec un cut-off à 169. Ce dernier correspondant à une sensibilité de 93 % et une spécificité de 86 % avec une excellente discrimination (aire sous la courbe ROC de 0,95-0,97) (14).
Si nous appliquons ces deux méthodes diagnostiques à notre exemple, la première exclut la présence d’un syndrome hémophagocytaire à deux reprises. Notre patient ne regroupant que 3 critères diagnostiques sur les 5 exigés au minimum lors de la première ponction médullaire (fièvre, bi-cytopénie et hyperferritinémie) contre 4 lors de la seconde ponction (pancytopénie, hyperferritinémie, hypertriglycéridémie/hypofibrinogénémie et hémopha-gocytose médullaire). En revanche, il présente un H-score de 142 soit une probabilité de 17 % lors de la première ponction qui se majore à 232 soit une probabilité de 98% d’être face à un syndrome hémophagocytaire lors de la seconde ponction. En outre, notre exemple souligne la nécessité de répéter les dosages et le caractère rapidement évolutif du SH.
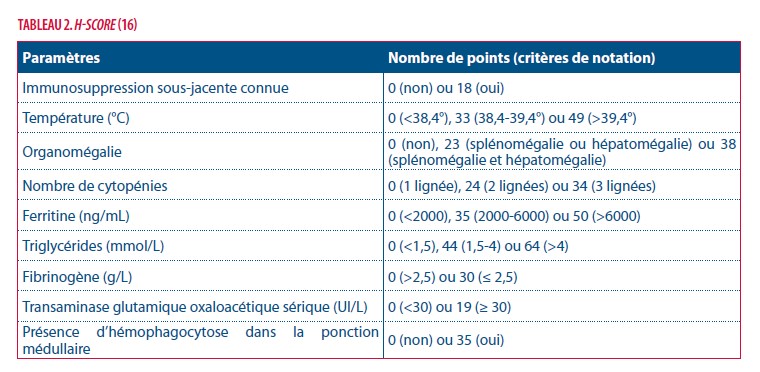
Parmi les critères diagnostiques communs à ces deux méthodes, l’hémophagocytose médullaire est le seul critère nécessitant la réalisation d’un acte invasif (la ponction médullaire) pour en confirmer le diagnostic. Dans la majorité des cas, cette dernière sera rapidement réalisée par le praticien face à une suspicion de SH. Néanmoins, cet examen n’est ni sensible (70-83 %) ni spécifique (60%). En d’autres termes, l’hémophagocytose n’est ni suffisante ni nécessaire au diagnostic de SH. Certaines situations cliniques étant à l’origine de faux positifs (ex. en cas de polytransfusion). En outre, elle peut être retardée parfois jusqu’à l’initiation du traitement d’où l’importance de répéter les médullogrammes comme l’illustre notre exemple (14).
Le traitement du SH comporte 3 grandes étapes. La première consiste en un traitement de soutien : transfusions selon la gravité des cytopénies, prise en charge des troubles de l’hémostase, … La deuxième consiste en un traitement étiologique. La dernière consiste en la suppression de l’augmentation de la réponse inflammatoire et au contrôle de la prolifération cellulaire (cellules NK et lymphocytes T cytotoxiques) en utilisant des agents immunosuppresseurs ou immunomodulateurs et des médicaments cytotoxiques si on observe une dégradation clinique du patient malgré le traitement étiologique. Parmi ces agents, seul l’étoposide a été validé pour son action efficace anti-lymphocytes T cytotoxiques (14). En ce qui concerne les SH associés à une infection virale autre que l’EBV, le traitement causal seul est suffisant et assure une guérison chez 60-70 % des patients (14,15).
L’aciclovir est le traitement de référence des infections à HSV1 à la dose de 10 mg/kg/8h en intraveineuse pour un minimum de 10 jours, durée à ajuster en fonction des évolutions cliniques et biologiques (9). Dans notre cas, la durée était de 18 jours compte tenu de l’amélioration lentement progressive des paramètres cliniques et biologiques de notre patient.
Conclusion
En conclusion, l’hépatite virale à HSV1 est une hépatite virale rare, potentiellement mortelle, pouvant être associée à un syndrome hémophagocytaire réactionnel. Son diagnostic reste néanmoins difficile compte tenu de la symptomatologie peu spécifique en début de la maladie. Elle survient plus fréquemment chez des personnes immunodéprimées, mais peut atteindre les immunocompétentes. Son évolution rapidement défavorable exige l’instauration d’un traitement précoce par aciclovir, ce dernier réduisant le taux de mortalité et le risque d’une transplantation hépatique.
Recommandations pratiques
- Face à une hépatite cytolytique sévère associée à des cytopénies, pensez à l’hépatite herpétique y compris chez les patients immunocompétents.
- Le syndrome hémophagocytaire réactionnel est une complication rare de l’hépatite herpétique qui en péjore le pronostic. Le H-score permet d’en déterminer le risque.
- L’initiation précoce du traitement est primordiale afin de diminuer le taux de mortalité et le risque de transplantation hépatique.
- Le traitement de référence de cette infection est l’aciclovir à la dose de 10 mg/kg/8h pour une durée minimale de 10 jours selon l’évolution clinique du malade.
Affiliations
(1) Cliniques universitaires Saint Luc – Université catholique de Louvain, Service de médecine interne et maladies infectieuses, B-1200 Bruxelles
(2) Centre Hospitalier du Luxembourg (Grand-Duché du Luxembourg), Service de maladies infectieuses, L-1210 Luxembourg
Correspondance
Dr Sarah Brilot
Cliniques universitaires Saint Luc - Université catholique de Louvain
Service de médecine interne et maladies infectieuses
Avenue Hippocrate 10, B-1200 Bruxelles
Sarah.brilot@gmail.com
Références
- Pathikonda M, Munoz SJ. Acute liver failure. Ann Hepatol. 2010 Jan-Mar;9(1):7-14.
- Then EO, Gayam V, Are VS, et al. Herpes Simplex Virus Hepatitis: A Brief Review of an Oft-overlooked Pathology. Cureus. 2019; 11(3): e4313.
- Organisation Mondiale de la Santé, Herpès (virus de l’herpès), 1 mai 2020. https://www.who.int/fr/news-room/fact-sheets/detail/herpes-simplex-virus
- James C, Harfouche M, Welton N, Turner K, Abu-Radddad L, Gottlieb S, Looker K. Herpes simplex virus : global infection prevalence and incidence estimates, 2016. Bull World Heath Organ. 2020 May 1 ; 98(5) : 315–329.
- Ajith Kumar AK, Magda D. Mendez. Herpes Simplex Encephalitis. StatPearls Publishing. February 8, 2021.
- Mailles A, Stahl JP. Infectious encephalitis in France in 2007: a national prospective study. Clin Infect Dis. 2009;49(12):1838-47.
- Patterson J, Hussey HS, Silal S, et al. Systematic review of the global epidemiology of viral-induced acute liver failure. BMJ Open. 2020;10:e037473.
- Norvell J, Blei A, Jovaconovic B, Levitsky J. Herpes simplex virus hepatitis: an analysis of the published literature and institutional cases. Liver transplantation. 2007 ; 13 :1428-1434.
- Mihalcea-Danciu M et al. Hépatite herpétique avec syndrome d’activation macrophagique chez un patient immunocompétent. La Revue de médecine interne. 2014 ; 35 : 823–826.
- Liew JW, Jones BL, Hunter AJ. Disseminated Herpes Simplex Masquerading as Hemophagocytic Lymphohistiocytosis : A case report. Perm j. 2019, 23 :18-202.
- Navaneethan U, Lancaster E, Venkatesh P, Wang J, Neff G. Herpes Simplex Virus Hepatitis - It’s High Time We Consider Empiric Treatment. JGLD [Internet]. 1Mar.2011 [cited 1Nov.2021];20(1):93-6.
- Rouphael NG, Talati NJ, Vaughan C, Cunningham K, Moreira R, Gould C, Infections associated with haemophagocytic syndrome. Lancet Infect Dis. 2007 ; 7 : 814-822.
- McClain KL, Eckstein O. Clinical features and diagnosis of hemophagocytic lymphohistiocytosis. In : UpToDate, Post TW (Ed), UpToDate, Waltham, MA. (Accessed on july 07, 2021.)
- Novotny F, Simonetta F, Samii K, Chalandon Y, Serratrice J. ‘Syndrome hémophagocytaire réactionnel’, Rev Med Suisse. 2017; volume 3. no. 579, 1797 – 1803
- Yabushita T, Yoshioka S, Koba Y, Ono Y, Hiramoto N, Tabata S, et al. Successful Treatment of Herpes Simplex Virus (HSV)-1-associated Hemophagocytic Lymphohistiocytosis (HLH) with Acyclovir: A Case Report and Literature Rev Intern Med (Tokyo, Japan). 2017; 56(21), 2919–2923.
- Fardet L, Galicier L, Lambotte O, Marzac C, Aumont C, Chahwan D et al. Development and validation of the HScore, a score for the diagnosis of reactive hemophagocytic syndrome. Arthritis Rheumatol. 2014 Sep;66(9):2613-20.