INTRODUCTION
L’hypercholestérolémie familiale (HF) est causée par la présence d’un (seul) allèle muté (état hétérozygote) des gènes LDLR, codant le LDL récepteur, APOB codant l’apolipopotéine B responsable de la liaison entre particules LDL et leur récepteur (1) ou PCSK9 codant pour la « Proprotéine convertase subtilisine/kexine de type 9 » qui facilite la dégradation lysosomiale des récepteurs aux LDL) (2) (Figure 1). Un seul allèle muté contribue à réduire de 50 % l’épuration hépatique des lipoprotéines LDL, produisant ainsi un taux de LDL-cholestérol (LDL-C) deux fois supérieur aux taux habituels depuis la naissance.

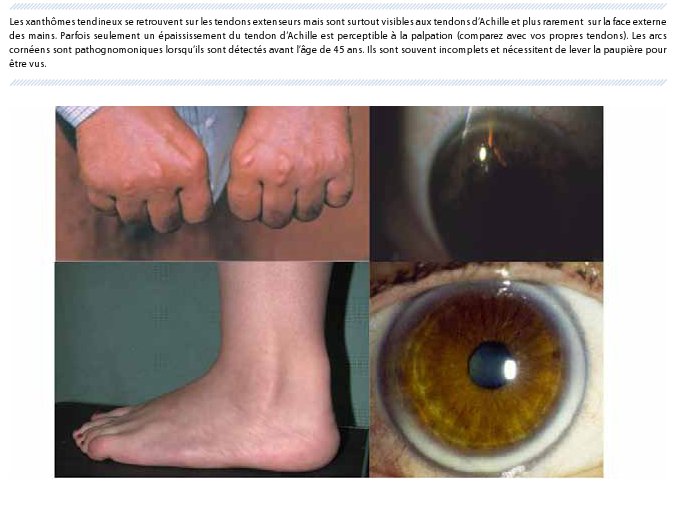
Le cholestérol se dépose ainsi dans les parois artérielles conduisant à des complications cardiovasculaires précoces. Ils se déposent aussi typiquement dans d’autres tissus produisant l’apparition d’arcs cornéens (ressemblant aux gérontoxons mais à un âge plus précoce) et des xanthomes tendineux (nodosités sur les tendons des mains ou sur les tendons d’Achille) (Figure 2). Étant donné le caractère autosomal dominant de la transmission de la maladie, d’autres membres de la famille présentent un tableau semblable à celui du patient.
Il existe aussi une forme homozygote où les deux copies de gènes sont anormales. Le système de capture est alors quasiment aboli et le taux de cholestérol LDL s’élève parfois jusqu’à 10 fois les valeurs habituelles, au point de produire des problèmes cardiaques dès l’enfance. Cette situation est cependant bien plus rare (1/1000.000, moins de 10 cas connus en Belgique) et nous n’en parlerons pas ici pour nous concentrer sur la forme hétérozygote, plus fréquente.
La prise en charge de l’hypercholestérolémie familiale a fait récemment l’objet de recommandations européennes pour la prise en charge chez l’enfant et l’adolescent (3) ainsi que l’adulte (4), suivant en cela la mise sur pied de réunions de consensus dans de nombreux pays, dont la Belgique (5).
Bien que l’HF et sa physiopathologie soient bien connues et bien que ses complications soient parables, elle reste une affection héréditaire encore largement sous-diagnostiquée et sous-traitée.
Cet article a pour but de sensibiliser le corps médical sur l’urgence de cette problématique et de développer des solutions pour la résoudre.
LES PROBLÈMES
L’hypercholestérolémie familiale (HF), bien que fréquente et sévère reste encore sous-diagnostiquée et sous-traitée.
2.1. Fréquence … Plus qu’on ne croit
Des études plus récentes (4) suggèrent de plus en plus l’idée qu’elle est bien plus répandue qu’on ne le pensait : 1/200 à 1/400. Au minimum, donc l’HF (Tableau 1) touche plus de 25.000 Belges. Ce serait donc la maladie génétique mortelle la plus fréquente.
2.2. Complications … désastreuses et prématurées
Ce taux de LDL-cholestérol très élevé (190-350 mg/dl) depuis la naissance (Tableau 1) produit des dépôts précoces et progressifs de cholestérol dans les parois artérielles et donc la survenue de maladies cardiovasculaires (MCV) entre 30 et 50 ans, même en l’absence de diabète, d’hypertension ou de tabagisme. Avant l’ère des statines, 40 % de ces hommes et 15 % de ces femmes en souffraient avant l’âge de 50 ans. Ceci donne une bonne idée du pronostic désastreux d’un patient qui ne serait pas traité à temps.

2.3. HF reste sous diagnostiquée et sous traitée en Belgique
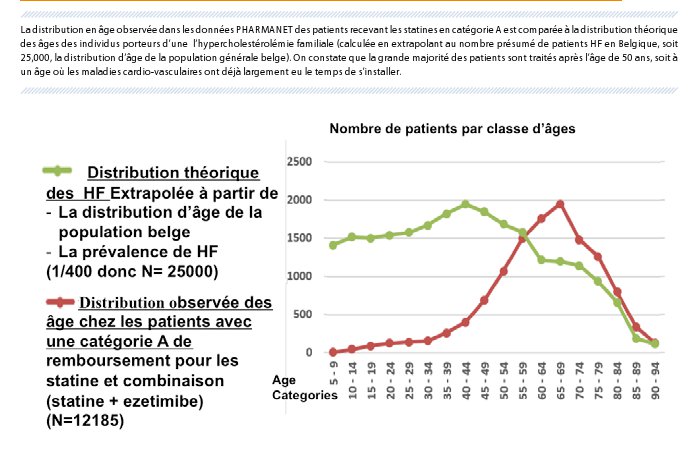
Il est difficile d’estimer le nombre de patients réellement diagnostiqués pour une HF en Belgique. Sur base des données PHARMANET de l’INAMI (données 2014), environ 13,000 patients bénéficient du remboursement en catégorie A, supposé spécifique pour l’HF, ce qui correspondrait à presque 50% du nombre d’individus souffrant d’HF en Belgique. Vu la non spécificité des critères de remboursement en catégorie A, Il est probable que ces patients n’aient pas tous de réelle HF. Autre fait, lorsqu’on observe la distribution des âges des patients qui reçoivent les statines en catégorie A (Figure 3), on voit que la grande majorité des patients sont traités après l’âge de 50 ans, soit à un âge où les maladies cardio-vasculaires ont déjà largement eu le temps de s’installer.
Figure 3. Données Pharmanet sur les remboursements en catégorie A des statines
LES SOLUTIONS
3.1. Un diagnostic … clinique et facile
Une HF devrait être suspectée face à un taux élevé de LDL-C ou face à des antécédents personnels ou familiaux de maladie cardiovasculaire (avant 55 ans chez les hommes et 60 ans chez les femmes) (Tableau 2).
Chez l’adulte, il est possible de poser le diagnostic avec plus de certitude sur base des critères du Dutch Lipid Clinic Network (Tableau 3), qui prennent en compte les taux de LDL-C, les antécédents personnels et/ou antécédents familiaux et les signes cliniques pouvant être présents chez certains patients HF. Il suffit d’additionner le score de chaque catégorie pour établir le diagnostic. Ainsi, un patient coronarien de 55 ans chez qui est découvert un LDL-C à 330 mg/dl ou un patient de 42 ans sans antécédent chez qui est découvert un LDL-C de 250 mg/dl et un arc cornéen (Figure 2) ont certainement une HF. Les xanthomes tendineux (Figure 2) sont parfois plus sensiblement détectés par échographie (épaisseur antéropostérieure de plus de 5,8 mm du tendon d’Achille) (6).

Chez l’enfant, les critères qui permettent d’établir un diagnostic formel sont présentés au tableau 4. Chez un enfant d’un parent avec une HF, simplement un taux de LDL-C > 130 mg/dl indique une forte probabilité d’HF (95 %) (Tableau 4). Dans ce cas, un test génétique est conseillé pour assurer la certitude d’un diagnostic qui va imposer un traitement médicamenteux pour la vie.
La démonstration d’une mutation fonctionnelle sur le gène du LDLR, APOB ou PCSK9 prouve de manière univoque l’existence d’une HF. A contrario, un test génétique négatif n’exclut pas une HF, car sa sensibilité est d’environ 80 %. Les prélèvements pour une analyse génétique se font sur un tube de sang classiquement réservé pour l’ « hémato » (10 ml, tube ‘EDTA’, bouchon violet)

3.2. HF, un diagnostic familial
En tant que maladie génétique dominante s’exprimant à l’état hétérozygote, le diagnostic d’HF (par test génétique positif ou simplement par critères cliniques) chez un patient doit motiver un dépistage familial extensif : la moitié de sa fratrie ou de sa descendance directe risque d’avoir le même problème (25 % chez les parents du 2ème degré, tels que cousins …). Y inclure les enfants est essentiel. Diagnostiquer l’HF dès l’enfance est important : plus le diagnostic est précoce, plus l’adhésion à ces règles thérapeutiques sera facile à obtenir des enfants et plus le pronostic vital sera proche de tout autre individu.
3.3. Traiter précocement
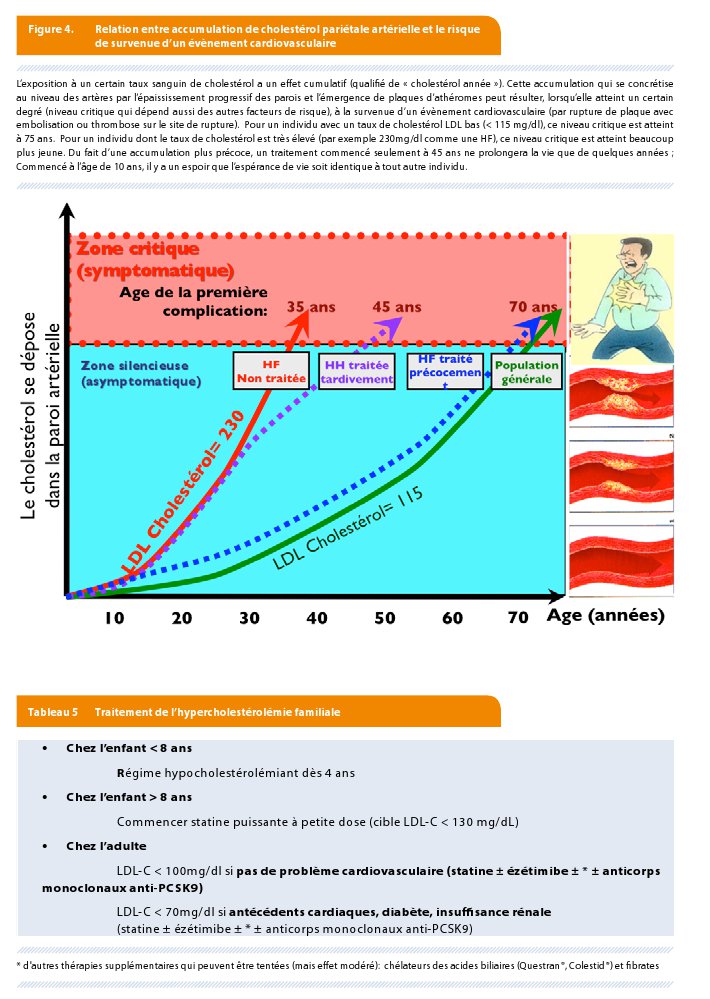
Le pronostic cardiovasculaire d’une personne avec une HF peut être amélioré et pourrait atteindre celui de toute autre personne si le taux de LDL-C est ramené au niveau des valeurs habituelles dès le plus jeune âge. L’explication est simple comme décrite à la Figure 4. Même si un bon équilibre diététique est impératif, normaliser les taux de LDL-C nécessitera la prise de médicament. Aucune étude randomisée contrôlée avec placebo n’a été effectuée, pour des raisons évidentes. Toutefois des études d’observation ont bien confirmé le bénéfice d’un traitement hypolipémiant chez les adultes (7) mais aussi chez les enfants (3).
3.3.1 Après 2 ans et avant 8 ans
Il est idéal de commencer un régime hypo-cholestérolémiant dès l’âge de 2 - 4 ans. Pas avant pour laisser le temps du développement cérébral qui nécessite un apport en graisses variées.
3.3.2 Chez l’enfant de plus de 8 ans
L’objectif sera de ramener le taux de LDL-C à des valeurs plus acceptables tout en le familiarisant avec la prise quotidienne d’un médicament (Tableau 5). Conduire un traitement médicamenteux aussi jeune (avant l’adolescence) facilitera l’adhésion thérapeutique future. Des exceptions tels que des taux très élevés de LDL-C (> 300 mg/dL chez des enfants hétérozygotes sévères ou chez enfants homozygotes), peuvent justifier la prescription avant 8 ans. La cible visée est un LDL-C en dessous de 130 mg/dL, ce que l’on obtiendra facilement à faible dose de statine mais que l’on choisira puissante (telle qu’atorvastatine 10 ou rosuvastatine 10) car ce seront celles-ci qu’il continuera de prendre à plus forte dose une fois adulte.
3.3.3 À l’âge adulte
Les cibles recommandées (Tableau 5) seront celles de la prévention de patients "à risque élevé" : la cible visée est un LDL-C en dessous de 100 mg/dl. Ceci nécessitera la prescription d’une statine puissante à bonne dose (associée à une réduction de plus de 50% du LDL-C) telle que atorvastatine 20 – 80 mg ou rosavastatine 10-40 mg souvent combinée à l’ézétimibe. En cas de complications cardiovasculaires, diabète ou insuffisance rénale, le taux de LDL-C devra être abaissé en-dessous de 70mg/dl (8, 9) (Tableau 5). Toutefois, étant donné le taux de départ très élevé du LDL-C, de telles cibles (70 ou même 100 mg/dL) peuvent être difficiles à atteindre. C'est la raison pour laquelle on se réjouit de l'arrivée de traitements adjuvants tels que les anticorps anti-PCSK9 (10). L’alirocumab (Praluent®) et l’évolocumab (Repatha®), en association avec une statine et l'ézétimibe permettent chez les patients HF d’atteindre moins de 100 ou même moins 70 mg/ dL chez une grande majorité de patients. L'alirocumab (PRALUENT®) est remboursé depuis septembre 2016 (Fiche pratique) chez des patients HF confirmé sur base d'un score DLCN > 8 en présence de taux trop élevés de LDL-C (>130 mg/dL ou même >100 mg/dL en cas d'antécédent de syndrome coronarien aigu).
3.4. www.belchol.be
Une association de patients pour l’hypercholestérolémie familiale s’est nouvellement formée : elle a pour but de sensibiliser le corps médical sur l’urgence de cette problématique. Elle a également pour objectif d’harmoniser le dépistage et le suivi des familles ainsi que de faciliter le remboursement des tests et des traitements. Belchol a déjà entrepris des actions au niveau du parlement européen (septembre 2015) et du parlement belge (novembre 2015). Sur le site (Figure 5, www.belchol.be, mais aussi sur Facebook et Twitter), le patient pourra trouver des informations sur cette maladie qui l’aideront, entre autre, à sensibiliser les membres de sa famille.
 3.5. Un remboursement spécial à adapter
3.5. Un remboursement spécial à adapter
Les traitements chez ces patients bénéficient d’un remboursement spécial en catégorie A comme certaines spécialités destinées au traitement du diabète ou du cancer. Ces critères de remboursement viennent d'être modifié en octobre 2016. En effet les anciens ne permettaient malheureusement pas le remboursement chez tous les patients avec une HF, particulièrement les enfants. Ces nouvelles conditions de remboursement apportent un progrès indéniable dans le remboursement du traitement des enfants souffrant d'hypercholestérolémie familiale. Elles permettent aussi de mieux définir la population des patients avec une hypercholestérolémie familiale traités en Belgique, tout en permettant la poursuite et l'initiation de traitement de patients à haut risque parce que porteur d'une autre dyslipidémie génétique que l'HF (Fiche pratique).
3.6. Des alertes aussi
Belchol, le Belgian Atherosclerosis Society/Belgian Lipid Club, la Belgian Society of Chemistry, et l’Institut Scientifique de Santé Publique ont récemment recommandé à tous les laboratoires d'attirer l’attention des praticiens par une alerte sur la possibilité d’une hypercholestérolémie familiale dès qu’un taux de LDL-C est anormalement élevé. Cette alerte encouragera le médecin à initier un dépistage de l’HF.
3.7. Projet de dépistage en cascade
Plusieurs expériences sont en cours (« Projet Koala-Lou » dans la région autour de La Louvière et « projet Bel-Cascade » en Wallonie et en Flandre) pour examiner la faisabilité de programmes de « dépistage en cascade ». L’appellation « cascade » fait référence au fait que, à partir d’une personne chez qui le diagnostic a déjà été confirmé, on peut en retrouver de nombreuses autres susceptibles de présenter la même maladie. Concrètement, le patient connu pour avoir une hypercholestérolémie familiale est informé de cette démarche. Si le patient consent, il rencontre une infirmière qui réalise avec lui l’arbre généalogique de sa famille. Par la suite, elle contacte les autres membres de la famille. Dès qu’une personne entre dans le programme, le médecin généraliste en est informé, afin d’assurer une meilleure prise en charge diagnostique et thérapeutique. Celui-ci peut aussi disposer de soutien via des conseils spécialisés.
CONCLUSIONS
L’hypercholestérolémie familiale (HF) fait partie de ces paradoxes de la médecine où une maladie continue de produire des complications dévastatrices, alors qu'elle est traitable, parce qu'elle reste sous-diagnostiquée. Elle n’est pas un simple désordre du cholestérol ou une entité génétique exotique, c’est avant tout un désastre familial où des membres de ces familles continuent de souffrir de maladies cardio-vasculaires, voire décèdent parfois très jeunes, dès l’âge de 30 ans. Et cela de génération en génération, jusqu’à ce que le médecin de famille pense au facteur génétique et donc à l’hypercholestérolémie familiale. La participation de tous (association de patients, sociétés scientifiques, médecins spécialistes et généralistes, pharmaciens, INAMI, mutuelles) sera nécessaire pour relever le défi de soigner tous les patients souffrant de cette maladie en Belgique.
affiliations
1 Président de Belchol (Association Belge de Patients pour l’Hypercholestérolémie Familiale www.belchol.be) Vice-président de la Société Belge d’Athérosclérose
Correspondance
Dr. Olivier S Descamps
Centres Hospitaliers Jolimont - Département de Médecine Interne 7100 Haine Saint-Paul -
Tel 064/23 31 67
Cliniques universitaires Saint-Luc Service de cardiologie B-1200 Bruxelles.
Tel 02/764 2812
olivierdescamps@hotmail.com
Références
-
Goldstein JL, Hobbs HH, Brown MS. Familial hypercholesterolemia. In: Scriver CR, Sly WS, Childs B, et al, eds. The metabolic and molecular bases of inherited disease. 8th ed. New York, NY: McGraw-Hill Companies Inc 2001.
-
Seidah NG, Benjannet S, Wickham L, Jasmin SB, Stifani S, Basak A, et al. The secretory proprotein convertase neural apoptosis-regulated convertase 1 (NARC-1): liver regeneration and neuronal differentiation, Proc Natl Acad Sci U S A 2003;100:928–933.
ouvrir dans Pubmed -
Wiegman A, Gidding SS, Watts GF, Chapman MJ, Ginsberg HN, Cuchel M, et al.; for the European Atherosclerosis Society Consensus Panel. Familial hypercholesterolaemia in children and adolescents: gaining decades of life by optimizing detection and treatment. Eur Heart J 2015;36(36):2425-37.
ouvrir dans Pubmed -
Nordestgaard BG, Chapman MJ, Humphries SE, Ginsberg HN, Masana L, Descamps OS, et al ; for the European Atherosclerosis Society Consensus Panel. Familial hypercholesterolaemia is underdiagnosed and undertreated in the general population: guidance for clinicians to prevent coronary heart disease: Consensus Statement of the European Atherosclerosis Society. Eur Heart J 2013;34(45):3478-90a.
ouvrir dans Pubmed -
Descamps OS, Tenoutasse S, Stephenne X, Gies I, Beauloye V, Lebrethon MC, et al. Management of familial hypercholesterolemia in children and young adults: consensus paper developed by a panel of lipidologists, cardiologists, paediatricians, nutritionists, gastroenterologists, general practitioners and a patient organization. Atherosclerosis 2011;218(2):272-80.
ouvrir dans Pubmed -
Descamps OS, Hondekijn JC, Van Leuven F, Heller FR. The use of Achilles tendon ultrasonography for the diagnosis of familial hypercholesterolemia. Atherosclerosis 2001, 157 : 514-518.
ouvrir dans Pubmed -
Versmissen J1, Oosterveer DM, Yazdanpanah M, Defesche JC, Basart DC, Liem AH, et al .Efficacy of statins in familial hypercholesterolaemia: a long term cohort study. BMJ 2008;337:a2423.
ouvrir dans Pubmed -
Reiner Z, Catapano AL, De Backer G, Graham I, Taskinen MR, Wiklund O, et al. ESC/ EAS Guidelines for the management of dyslipidaemias: the Task Force for the management of dyslipidaemias of the European Society of Cardiology (ESC) and the European Atherosclerosis Society (EAS). Eur Heart J 2011;32(14):1769-818.
ouvrir dans Pubmed -
Descamps OS, De Backer G, Annemans L, Muls E, Scheen AJ. Belgian Atherosclerosis Society/ Belgian Lipid Club. Les nouvelles recommandations européennes pour le traitement des dyslipidémies en prévention cardiovasculaire. Rev Med Liège 2012; 67: 118-127.
ouvrir dans Pubmed -
Descamps OS. Les inhibiteurs du PCSK9 : une nouvelle classe d’hypolipémiants. Louvain Med 2016 ; 135 (5) : 291-297.