L’institut des Maladies rares aux Cliniques universitaires Saint-Luc
Marie-Cécile Nassogne1, Marie-Françoise Vincent2, Olivier Devuyst3
Cliniques universitaires Saint-Luc, 1Neurologie Pédiatrique et Maladies héréditaires du Métabolisme ; 2Professeur émérite ; 3Service de néphrologie
Introduction
Le concept de maladies rares a émergé aux États-Unis sous la pression d’associations de malades, à l’occasion de l’adoption, en 1983, d’un règlement particulier pour les médicaments destinés au traitement de maladies rares. Ce règlement incluait plusieurs incitations pour convaincre les industriels d’investir dans ces médicaments rares à condition que le marché potentiel soit inférieur à 200 000 malades aux États-Unis soit une prévalence de 86 personnes atteintes pour 100 000 individus (1). En 1999, l’Union européenne (UE) définit une maladie rare comme une maladie chronique ou potentiellement mortelle dont la prévalence est inférieure à 50/100 000 soit 1/2000 et qui nécessite un effort particulier pour développer un traitement (2).
On dénombre près de 7000 maladies rares dont plus de 50 % concernent les enfants et 70-80 % sont d’origine génétique. On estime qu’environ 3.5-5.9 % de la population mondiale souffre d’une maladie rare, soit 263–446 millions de patients dans le monde, 18-30 millions dans l’UE et pour la Belgique pas moins de 600 000 personnes seraient concernées par une maladie rare avec comme difficultés l’odyssée diagnostique, le fait que ce soit une maladie chronique et le peu de traitements disponibles
En 2008, une directive européenne encourage les pays à mettre en place un plan national pour les maladies rares. Un groupe de travail organisé par la Fondation Roi Baudouin élabore un plan national maladies rares pour la Belgique (3). Ce plan reprend différents domaines avec l’amélioration de l'accès aux diagnostics et aux informations pour le patient, l’optimisation des soins, l’amélioration de l’acquisition des connaissances et la gouvernance et la durabilité du plan. Plusieurs acteurs sont impliqués dont le Service Public Fédéral Santé Publique, l’INAMI, Radio Org, … et dans le décours, plusieurs arrêtés royaux sont publiés en 2014.L’institut des Maladies rares est créé aux Cliniques universitaires Saint-Luc et sera coordonné par le Professeur Marie-Françoise Vincent.
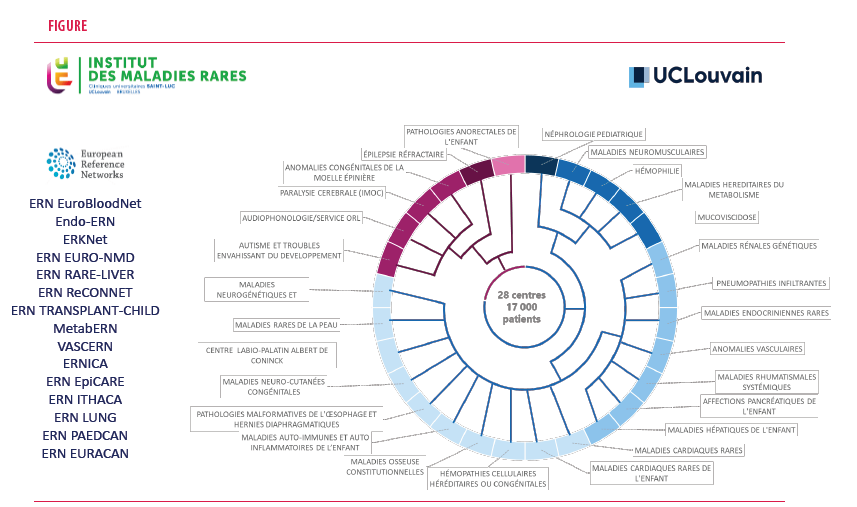
L’institut des Maladies rares des Cliniques universitaires Saint-Luc
Créé en 2014, l’Institut va se développer progressivement. En 2020, le Professeur Olivier Devuyst succède au Professeur Vincent à la coordination de l’Institut des Maladies rares. L’équipe se renforce avec la collaboration de Madame Fabienne Lohest qui assure un lien important avec les patients et les consultations multidisciplinaires, Madame Olivia Lacroix qui avec le Professeur Devuyst, coordonne les projets maladies rares et Madame Nissrine Douail, très impliquée dans la collecte des données pour les registres nationaux et internationaux. L’Institut regroupe 28 centres multidisciplinaires de prise en charge de patients souffrant de maladies rares avec près de 17 000 patients suivis. 15 équipes sont associées aux Réseaux Européens de référence ERNs (Les réseaux européens de référence –ERN - sont des réseaux virtuels réunissant des prestataires de soins de santé de toute l’Europe. Ils ont pour objectif de faciliter les échanges sur des maladies complexes ou rares ou des affections qui nécessitent un traitement hautement spécialisé et une concentration des connaissances et des ressources). Plus de 300 personnes sont impliquées avec des médecins mais aussi une série d’autres métiers dont l’aide est précieuse dans la prise en charge des patients.
Contacts
http://www.institutdesmaladiesrares.be/
L’Institut a pour souhait d’aider au mieux les patients atteints de maladies rares et d’optimaliser leur prise en charge.
Téléphone : 02 764 78 00.
Permanence téléphonique : Mardi de 9h à 12h & Jeudi de 13h à 16h.
maladies.rares@saintluc.uclouvain.be
Références
- Reagan, Ronald W. (January 4, 1983). "Statement on Signing the Orphan Drug Act - January 4, 1983". Internet Archive. Washington, D.C.: National Archives and Records Service. pp. 9–10.
- Règlement (CE) no 141/2000 du Parlement européen et du Conseil du 16 décembre 1999 concernant les médicaments orphelins (JO L 18 du 22.1.2000, p. 1-5)
- https://www.health.belgium.be/sites/default/files/uploads/fields/fpsheal...
Le dépistage néonatal de l’amyotrophie spinale et les nouvelles thérapies associées
Joseph P. Dewulf
Laboratoire des Maladies Métaboliques Héréditaires (Biochimie Génétique) et Centre de Dépistage Néonatal, Service de Biochimie Médicale, Cliniques universitaires Saint-Luc, UCLouvain, joseph.dewulf@saintluc.uclouvain.be
L’amyotrophie spinale
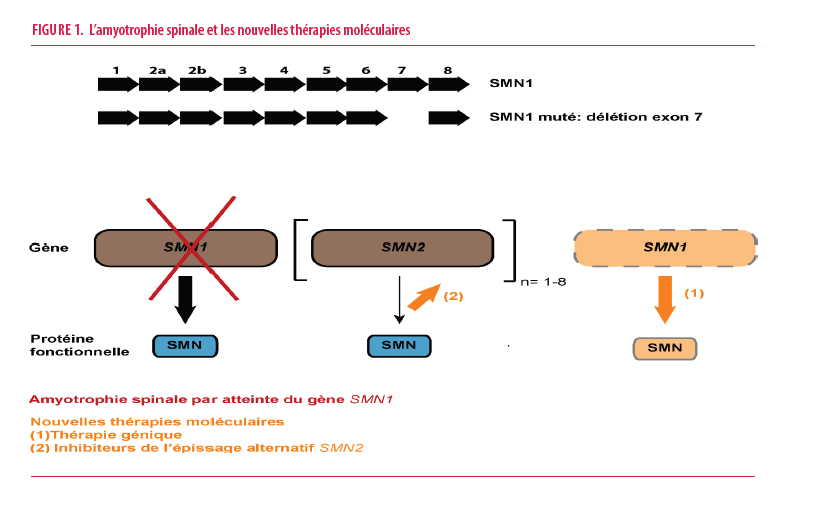
L’amyotrophie spinale est une maladie neuromusculaire rare d’origine génétique autosomique récessive, entrainant une dégénérescence des motoneurones de la moelle épinière et par conséquent une paralysie musculaire progressive et irréversible. En l’absence de ventilation mécanique, le décès était inexorable avant l’âge de deux ans dans la forme classique de la maladie (maladie de Werdnig-Hoffmann ou amyotrophie spinale de type 1). D’un point de vue génétique, la grande majorité des patients atteints (95%) est homozygote pour une délétion de l’exon 7 du gène SMN1 entrainant un défaut de protéine SMN fonctionnelle (Figure 1). La protéine SMN joue un rôle physiologique important dans l’épissage des ARN pré-messagers, nécessaire à la survie des motoneurones de la corne antérieure de la moelle épinière. L’incidence à la naissance de cette maladie varie entre 1/7000 et 1/28 000 en Occident selon les séries rapportées. Il existe un gène paralogue de SMN1, appelé SMN2, codant également pour la protéine SMN. Cependant, seulement 10 % des protéines SMN issues de SMN2 sont fonctionnelles en raison d’un épissage alternatif de son exon 7, lié à la présence d’un polymorphisme particulier. Le nombre de copies du gène SMN2 varie entre 1 et 8 copies chez les individus et en cas d’amyotrophie spinale, la sévérité de la maladie sera inversement proportionnelle à ce nombre de copies.
Révolutions thérapeutiques
Récemment, trois thérapies moléculaires de nouvelle génération, acceptées par la FDA et l’EMA en tant que médicaments orphelins entre 2016 et 2021, ont montré des résultats exceptionnels sur le développement moteur des patients atteints d’amyotrophie spinale, en particulier si le traitement est commencé au stade pré-symptomatique. Une de ces thérapies repose sur une approche de thérapie génique par injection intraveineuse d’une dose d’adénovirus atténué modifié contenant le gène fonctionnel SMN1 (Figure 1). Les deux autres thérapies approuvées à l’heure actuelle reposent sur l’utilisation de molécules thérapeutiques visant à réduire l’épissage alternatif de SMN2 afin d’élever la proportion de protéine SMN fonctionnelle issue de ce gène paralogue de SMN1 (Figure 1). Ces résultats thérapeutiques ont justifié l’intérêt pour un dépistage néonatal systématique ciblé. Après avoir fait l'objet d'une étude pilote, ce dernier a été ajouté au programme officiel du dépistage néonatal de la Fédération Wallonie-Bruxelles en mars 2021. Le test repose sur une amplification qPCR ciblant le gène SMN1 afin de détecter une homozygotie de la délétion exon 7 à partir d’ADN génomique extrait d'un spot de sang séché.
Le dépistage néonatal sur sang séché en bref
Tous les ans, quelques dizaines d’enfants atteints de maladies rares sont détectés parmi les 120 000 nouveau-nés en Belgique grâce aux programmes de dépistage néonatal financés par les régions. Les cinq centres de dépistage agréés en Belgique réalisent des tests de laboratoire ciblés au départ de sang séché récolté à la naissance (entre 2 et 4 jours de vie) sur papier buvard (les « cartes de Guthrie »). Toutes les maladies recherchées sont des maladies pour lesquelles un traitement ou une prise en charge adaptée et précoce entrainera une amélioration significative du pronostic. Face à l’évolution des connaissances, des thérapies disponibles, et des tests de dépistage, la liste des maladies candidates au dépistage néonatal s’allonge régulièrement. En 2022, le programme officiel permet de dépister 19 maladies rares (Table 1, maladies classées par ordre d'incidence estimée de haut en bas, actualisé en mars 2021, la plus fréquente étant l'hypothyroïdie congénitale avec une incidence de 1/2500 nouveau-nés). A l’exception de la majorité des cas d’hypothyroïdie congénitale, toutes les maladies dépistées sont d’origine génétique de transmission autosomique récessive.

Dr Warehouse – Accélérer la recherche translationnelle pour les maladies rares
Nicolas Garcelon
Institut Imagine, Dr Warehouse
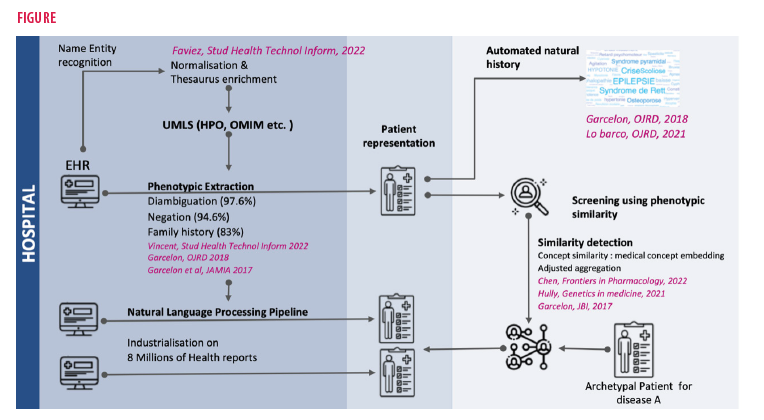
En amont des registres sur les maladies rares, nous pensons qu’il faut accélérer la réutilisation des données hospitalières, saisies au cours du soin, pour permettre aux chercheurs et aux médecins de rapidement fouiller la mémoire collective de l’hôpital. A cette fin, nous avons conçu Dr Warehouse, un entrepôt de données hospitalier, intégrant les données structurées (biologie) et non structurées (comptes rendus). Adossé à ces données, nous avons développé un moteur de recherche multimodal, permettant de retrouver des patients à partir des données contenues dans le texte libre et/ou les données structurées (Garcelon, 2018). Il permet d’accélérer l’inclusion des patients dans des études, mais aussi de réaliser des tests de faisabilité, des tests d’hypothèse d’association de signes, ou seulement de retrouver des patients utiles pour prendre en charge un nouveau patient au cours d’une réunion de concertation pluridisciplinaire.
Afin de faciliter l’extraction et l’analyse des données des patients, nous avons développé un algorithme d’extraction automatique des phénotypes à partir des comptes rendus (Garcelon, 2018, Vincent 2022). A partir de cette restructuration des données issues du texte libre, nous avons créé un algorithme de similarité qui calcule une distance phénotypique entre les patients (Garcelon, 2017 ; Chen 2022). Nous l’avons utilisé pour retrouver des patients en errance diagnostique de maladies rares, par exemple sur le gène KCNA2 (Hully, 2021).
Plus de 54 publications à Necker ont utilisé Dr Warehouse pour réaliser leur recherche.
Dr Warehouse est open source (Garcelon, JBI, 2018), et est installé dans plusieurs hôpitaux en France. Notre nouvel objectif est d’utiliser ces algorithmes de transformation du dossier patient et de détection de patients en errance dans des hôpitaux généralistes pour lesquels le risque d’errance est plus important.
Les thérapies par ARN interférents : une révolution thérapeutique en marche pour les maladies rares et moins rares
Arnaud Devresse, MD, PhD1, Valentine Gillion, MD1
Cliniques universitaires Saint-Luc 1Département de Néphrologie, B-1200 Bruxelles, Belgique
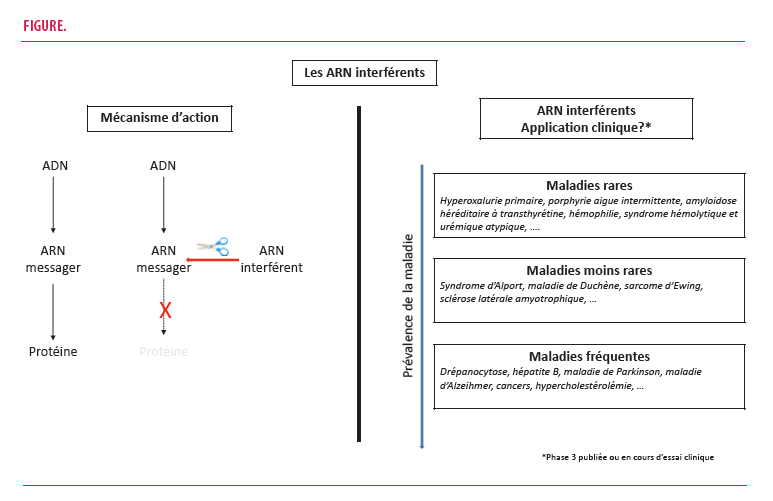
L’ARN interférence (ARNi) est un mécanisme cellulaire naturel permettant la régulation de l’expression d’un gène via des « small interfering RNAs » (siRNAs). Des siRNAs synthétiques peuvent être développés afin de cibler un transcrit d’ARN messager endogène d’un gène donné, conduisant à son clivage et in fine à la suppression de la synthèse de la protéine encodée (1).
Depuis les premiers essais pré-cliniques à la fin du XXème siècle, nous assistons à une véritable explosion des applications potentielles plus ou moins avancées dans leur développement clinique (2). Dans le domaine des maladies rares, les médicaments ARNi ont été - ou sont actuellement - testés dans de plusieurs affections génétiques, dont l’amyloidose héréditaire à transthyrétine (ATTR), la porphyrie aigue intermittente (PAI), et l’hyperoxalurie primaire de type 1 (HP1), que nous allons détailler (2).
L’ATTR est une maladie autosomique dominante rare causée par une mutation génétique dans le gène de la transthyrétine TTR. Il en résulte la formation de fibrilles amyloides à TTR qui vont se déposer dans différents organes, entrainant leur dysfonction. L’ATTR se manifeste essentiellement par des atteintes des systèmes nerveux autonome et périphérique, du cœur, et du tractus digestif. Le patisiran est un traitement ARNi qui inhibe la formation hépatique de la TTR mutée. Une étude de phase 3 (APPOLO) a démontré l’efficacité et la sécurité du patisiran afin de : a). diminuer de manière rapide et durable les concentrations sériques de TTR ; b). améliorer de manière significative la symptomatologie clinique, notamment neurologique, des patients atteints d’ATTR (3).
Les prophyries sont un groupe de maladies provoquées par la carence en enzymes impliquées dans la synthèse de l’hème. La PAI est la plus fréquente des prophyries aigues. Il s’agit d’une maladie autosomique dominante qui est secondaire à une carence en enzyme porphobilinogène désaminase (appelée également hydroxyméthylbilane synthase), qui provoque une accumulation des précurseurs des porphyrines acide delta-aminolévulinique (ALA) et porphobilinogène (PBG) initialement limitée au foie. Il en résulte une symptomatologie évoluant en crises avec des manifestations peu spécifiques, essentiellement des systèmes nerveux central (confusion, anxiété, dépression, hallucinations, épilepsie,..), autonome (douleur abdominale, constipation, tachycardie,…), et périphérique (paresthésie, douleur, paralysie, ..). Le givosiran est un traitement ARNi qui inhibe la production hépatique d’ALA/PBG. Le givosiran a démontré sa sécurité et son efficacité dans une étude de phase 3 afin de : a). diminuer de manière rapide et durable les concentrations sériques d’ALA ; b). améliorer la symptomatologie clinique des patients souffrant de PAI (4).
L’HP1 est une maladie autosomique récessive très rare, causée par la dysfonction de l’enzyme hépatique alanine-glyoxylate aminotransférase (AGT) qui catalyse la transamination du glyoxylate en glycine. Il en résulte une production excessive d’oxalate et de glycolate. L’oxalate forme ensuite un sel calcique hautement insoluble. Le seul organe capable d’éliminer cet excès d’oxalate est le rein. À un stade précoce, l’HP1 se manifeste cliniquement par des épisodes répétés de lithiases urinaires, par de la néphrocalcinose et finalement par un déclin progressif et inéluctable de la fonction rénale. Le lumasiran est un traitement ARNi qui inhibe la glycolate oxydase (GO) hépatique (5). Il en résulte une déviation de la production d’oxalate vers la production de glycolate qui est inoffensive pour le rein. Le lumasiran a démontré sa sécurité et son efficacité afin de diminuer de manière rapide et durable la production hépatique d’oxalate (6-8). L’impact du lumasiran sur l’outcome clinique des patients avec PH1 est en cours d’investigation.
Les ARNi sont également actuellement testés dans plusieurs autres maladies moins rares, voire fréquentes (2). Par exemple, l’inclisiran est un ARNi qui inhibe la formation de PCSK9, ce qui conduit à une diminution de la dégradation lysosomale du récepteur au LDL cholestérol et donc au final, à une augmentation de la clairance du LDL circulant (9). Des données cliniques préliminaires ont montré l’efficacité de l’inclisiran pour diminuer de manière importante et durable les concentrations sériques de LDL cholestérol. De plus, cet effet spectaculaire s’observait également chez les patients traités par statines au moment de l’inclusion dans l’étude (10). Depuis lors, une multitude d’essais cliniques sont en cours pour valider sur de larges cohortes ces observations et pour déterminer l’impact de l’inclisiran sur l’outcome clinique cardiovasculaire (2).
Les ARNi ouvrent donc un nouveau chapitre important dans le traitement de nombreuses maladies, rares et moins rares. Néanmoins, plusieurs questions doivent encore être résolues. Parmi celles-ci, leur accès, notamment dans les pays en voie de développement, sera un point critique. En effet, ces nouveaux traitements, aussi prometteurs soient-ils, ont un coût important. La communauté médicale, au sens large du terme, devra rester vigilante afin qu’aucun patient, notamment porteur de maladie rare, ne soit exclu de cette avancée scientifique importante.
Conflit d’intérêt : AD, scientific advisory board pour Alnylam.
Financement : aucun
Références
- Milliner DS. siRNA Therapeutics for Primary Hyperoxaluria: A Beginning. Mol Ther. 2016; 24 (4): 666-667.
- Setten RL, Rossi JJ, Han SP. The current state and future directions of RNAi-based therapeutics. Nat Rev Drug Discov. 2019;18(6):421-446.
- Adams D, Gonzalez-Duarte A, O'Riordan WD, Yang CC, Ueda M, Kristen AV, et al. Patisiran, an RNAi Therapeutic, for Hereditary Transthyretin Amyloidosis. N Engl J Med. 2018; 379(1):11-21.
- Balwani M, Sardh E, Ventura P, Peiró PA, Rees DC, Stölzel U, et al. Phase 3 Trial of RNAi Therapeutic Givosiran for Acute Intermittent Porphyria. N Engl J Med. 2020; 382(24):2289-2301.
- Devresse A, Cochat P, Godefroid N, Kanaan N. Transplantation for primary hyperoxaluria type 1: designing new strategies in the era of promising therapeutic perspectives. Kidney Int Rep. 2020; 5 (12): 2136-2145.
- Garrelfs SF, Frishberg Y, Hulton SA, Koren MJ, O'Riordan WD, Cochat P, et al. Lumasiran, an RNAi Therapeutic for Primary Hyperoxaluria Type 1. N Engl J Med. 2021; 384(13):1216-1226.
- Sas DJ, Magen D , Hayes W , Shasha-Lavsky H , Michael M, Schulte I, et al. Phase 3 trial of lumasiran for primary hyperoxaluria type 1: A new RNAi therapeutic in infants and young children. Genet Med. 2022; 24(3):654-662.
- Mini Michael, J. G., Hadas Shasha-Lavsky, John C. Lieske, Yaacov Frishberg, Eva Simkova, Anne-Laure A. Sellier-Leclerc, Arnaud Devresse, Fitsum Guebre Egziabher, Sevcan A. Bakkaloglu, Chebl Mourani, Rola Saqan, Richard F. Singer, Richard G. Willey, Bahru A. Habtemarian, Ishir Bhan, Tracy McGregor, Daniella Magen. ILLUMINATE-C, a Single-Arm, Phase 3 Study of Lumasiran in Patients with Primary Hyperoxaluria Type 1 and CKD Stages 3b-5, Including Those on Hemodialysis. Presented at ASN Kidney Week 2021 (2021).
- Seidah NG, Prat A. The Multifaceted Biology of PCSK9. Endocr Rev. 2022; 43(3):558-582.
- Fitzgerald K, White S, Borodovsky A, Bettencourt BR, Strahs A, Clausen V, et al. A Highly Durable RNAi Therapeutic Inhibitor of PCSK9. N Engl J Med. 2017; 376(1):41-51.
La révolution thérapeutique de l’hémophilie
Cédric Hermans
Cliniques universitaires Saint-Luc, Unité d'Hémostase - Thrombose/hémophilie, B-1200 Bruxelles
Conséquence d’un déficit de production par le foie du facteur VIII (FVIII) ou IX (FIX) de la coagulation sanguine, l’hémophilie se caractérise par des hémorragies profondes qui affectent essentiellement les articulations et les muscles. Survenant spontanément chez les patients sévèrement atteints (FVIII et FIX < 1 %), ces hémorragies sont responsables d’une destruction des articulations et d’une arthropathie douloureuse et invalidante.
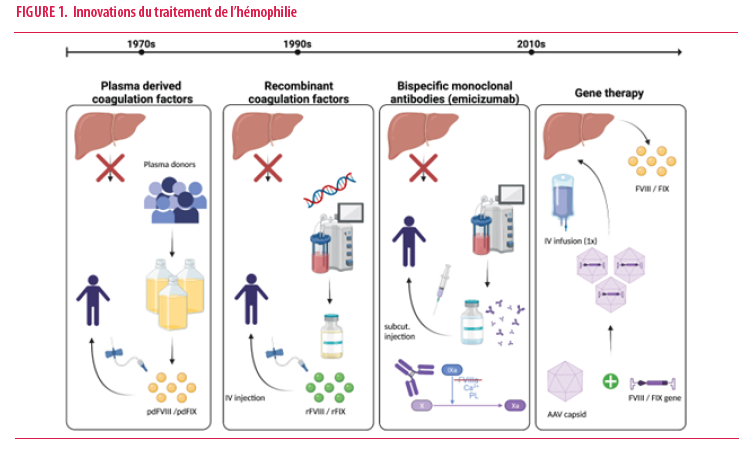
Le traitement « classique » de l’hémophilie est une thérapie dite de substitution dont le but est de maintenir chez les patients hémophiles sévères une quantité minimale de FVIII ou FIX dans le sang afin d’empêcher les hémorragies spontanées. Cette thérapie a évolué au cours du siècle dernier faisant appel initialement à du sang total, puis à du plasma et finalement des concentrés de FVIII et FIX d’origine plasmatique (Figure 1).
Ces premiers succès thérapeutiques ont été ternis par le drame de la contamination de milliers de patients hémophiles par les virus HIV et de l’hépatite C. L’introduction de techniques de sécurisation des concentrés de facteurs plasmatiques et surtout le développement de concentrés de FVIII et FIX synthétiques produits par biotechnologie, rendu possible par le clonage des gènes du FVIII et FIX, ont permis aux patients d’avoir accès à des traitements dénués de risques infectieux. La nécessité d’administrer ces concentrés de facteurs par voie intraveineuse, leur courte demi-vie plasmatique justifiant des traitements fréquents (jusqu’à plusieurs fois par semaine), leurs concentrations fluctuantes et leur immunogénicité se soldant chez certains patients par le développement d’anticorps neutralisants, représentent autant d’obstacles au traitement efficace et bien toléré de l’hémophilie.
De multiples innovations thérapeutiques au cours des deux dernières décennies ont révolutionné la prise en charge des patients hémophiles. Il s’agit de concentrés de facteurs synthétiques produits par biotechnologie et dotés d’une demi-vie prolongée (Figure 1). Obtenus par fusion avec l’albumine ou le fragment Fc des immunoglobulines ainsi que par pégylation, les concentrés à demi-vie prolongée réduisent le nombre d’administrations intraveineuses.
Leurs avantages sont indéniables parmi les patients hémophiles B chez lesquels les concentrés de FIX à demi-vie prolongée sont administrés toutes les semaines voire toutes les 3 semaines alors que pour les patients hémophiles A, les concentrés de FVIII à demi-vie prolongée doivent être en moyenne administrés 2 fois par semaine. Le développement d’un FVIII modifié à demi-vie ultra-prolongé, fusionné au fragment Fc des immunoglobulines et « découplé » du facteur von Willebrand endogène (BIVV001), nourrit l’espoir d’un traitement intraveineux une fois par semaine.
Pour les patients hémophiles A, la plus grande innovation est sans conteste le développement d’un anticorps bispécifique capable de mimer l’action du FVIII au sein de la cascade de la coagulation. Cet anticorps (emicizumab / Hemlibra®) met en contact le FIXa et le FX de la coagulation et joue le rôle de catalyseur de la coagulation assumé normalement par le FVIII (Figure 1).
Administré par voie sous-cutanée, doté d’une longue demi-vie, non reconnu par les anticorps dirigés contre le FVIII présents chez certains patients, ce traitement révolutionne le traitement de l’hémophilie A. Il permet en effet de maintenir une activité hémostatique constante équivalente à celle rencontrée chez les patients hémophiles A dits mineurs (FVIII proche de 15 %) moyennant des injections sous-cutanées hebdomadaires ou mensuelles. Cet anticorps bispécifique s’impose de plus en plus comme le traitement de référence des patients hémophiles A sévères et modérés. Ces derniers n’ont recours à des injections intraveineuses adjuvantes de FVIII qu’en cas d’accident ou de geste invasif justifiant une normalisation de la coagulation (FVIII > 50 %).
Une autre approche innovante très originale consiste à agir sur l’équilibre délicat de la cascade de la coagulation en réduisant les concentrations ou en inhibant les freins endogènes de la coagulation. Il s’agit principalement de l’antithrombine et du TFPI (Tissue Factor Pathway Inhibitor). Soumise à moins d’inhibition, la coagulation des patients hémophiles traités par ces « rebalancing agents » est davantage active. Ces traitements s’avèrent sans distinction efficaces chez les patients avec déficit en FVIII ou FIX, avec ou sans anticorps neutralisants. Les résultats des essais cliniques en cours sont prometteurs. Des cas de thromboses ont toutefois été rapportés et préoccupent la communauté.
Finalement, ces dernières années ont démontré la possibilité de guérir l’hémophilie par la thérapie génique (Figure 1). Il s’agit d’introduire dans les hépatocytes les gènes des FVIII ou FIX véhiculés jusqu’au foie par des vecteurs viraux dotés d’un tropisme hépatique. Ces vecteurs sont administrés aux candidats en une seule administration intraveineuse. Cette approche a démontré sa faisabilité et son succès chez certains patients, surtout ceux qui présentent un déficit en FIX. La variabilité de la réponse totalement imprévisible, une réaction hépatique parfois sévère justifiant le recours à une immunosuppression parfois longue, des risques d’intégration dans le génome qui ne sont pas totalement exclus, représentent, parmi d’autres, des obstacles à une banalisation de cette approche très prometteuse.
Ces multiples innovations changent fondamentalement la prise en charge et les perspectives d’épanouissement de chaque patient hémophile. Le grand défi est d’identifier pour chaque patient l’option thérapeutique la plus appropriée, qu’elle soit validée ou en cours d’étude. Il s’agit aussi et surtout de faire en sorte qu’un nombre croissant de patients hémophiles à travers le monde ait accès à ces traitements efficaces et plus faciles à utiliser.
Mucoviscidose : un nouveau souffle avec les modulateurs du CFTR
Sophie Gohy, Silvia Berardis, Christophe Goubau
Cliniques universitaires Saint-Luc, Service de Pédiatrie spécialisée - Secteur de pneumologie pédiatrique et mucoviscidose
Décrite pour la première fois en 1938 par le Dr Dorothy Andersen, la mucoviscidose est la maladie létale génétique (autosomique récessive) la plus fréquente dans nos populations caucasiennes. En Belgique, elle touche un nouveau-né sur 2850. La protéine déficiente (CFTR) code normalement pour un canal à anions, principalement chlorures, qui est exprimé au pôle apical de nombreuses muqueuses du corps humain, ce qui fait de la mucoviscidose une maladie systémique. Néanmoins, l’atteinte digestive et pulmonaire dominent la clinique et cette dernière, conditionne le pronostic de la maladie. Pendant près de 70 ans, les traitements disponibles étaient symptomatiques et ont permis au fils des années d’augmenter considérablement l’espérance de vie. Pour un enfant né entre 2016 et 2020, la médiane de survie prédite est de 50 ans (en ne prenant pas en compte le potentiel impact sur le taux de mortalité des améliorations de la prise en charge clinique, notamment liées à l’avènement des modulateurs du CFTR) (1). Administrés par voie orale, les traitements modulateurs du CFTR permettent partiellement de corriger le défaut protéique et de restaurer la fonction de la protéine CFTR. Plusieurs modulateurs sont actuellement disponibles en fonction du type de mutations au sein du gène CFTR. En 2016, les patients belges détenant une mutation de classe 3 (mutations malheureusement peu fréquentes) ont eu accès à l’ivacaftor, un potentialisateur qui augmente la probabilité d’ouverture du canal CFTR (2). L’année passée, en 2021, les patients homozygotes pour la mutation F508del et hétérozygotes (F508del/mutations à fonction résiduelle) ont pu bénéficier de l’association d’ivacaftor et d’un correcteur (le lumacaftor pour les enfants de 2 à 11 ans homozygotes F508del et le tezacaftor pour les patients à partir de 12 ans homozygotes ou hétérozygotes) (3-5). Enfin, cette année, nous espérons avoir accès en Belgique à la trithérapie, elexacaftor, tezacaftor et ivacaftor (2 correcteurs et 1 potentialisateur) pour tous les patients porteurs d’au moins une mutation F508del (6-8). Les conditions exactes de remboursement ne sont pas encore connues (Figure 1).

La mutation F508del étant la plus fréquente, environ 85% des patients pourraient ainsi avoir accès à ce traitement dit hautement efficace. Les marqueurs diagnostics et pronostics de la maladie s’améliorent sous ce traitement : diminution des taux de chlorure dans la sueur (à des taux sous le seuil diagnostic de la maladie), amélioration de la fonction respiratoire, du BMI et de la qualité de vie, diminution des exacerbations, des hospitalisations. Et plus encore…
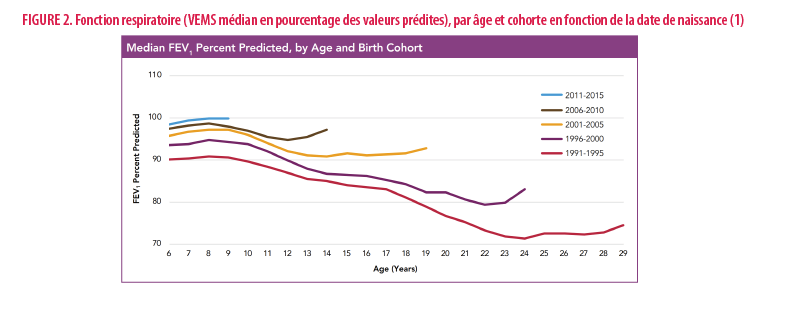
Les données du registre américain de 2020 sont particulièrement encourageantes et montrent une amélioration sans précédent de la fonction respiratoire des patients, et ce, dans toutes les tranches d’âge (Figure 2). Malgré un nombre moins important de spirométries réalisées, la réalisation de spirométries à domicile et la moindre mesure de la taille des enfants lors du suivi à distance en 2020 (en conséquence de la pandémie à coronavirus), beaucoup d’espoir ressort de ces données (1). Les études en vie réelle sur l’ivacaftor issues des registres américains et anglais montrent par ailleurs une réduction du risque annuel de mortalité et de transplantation d’organes (9). Similairement, après seulement 3 mois avec la triple thérapie en France, il y a une moindre proportion de patients en chemin vers la greffe pulmonaire (10).
En conclusion, nous vivons une période charnière dans le traitement des maladies rares, notamment la mucoviscidose. Des traitements particulièrement efficaces voient le jour. Ils changent réellement la vie de nos patients mais malheureusement à un coût prohibitif. Nous ne savons pas à quel point notre société sera capable d’assumer ces montants exorbitants et nous avons une chance énorme d’y avoir accès progressivement mais le combat pour un accès universel ne fait que commencer.
Références
- Cystic Fibrosis Foundation Patient Registry 2020 Annual Data Report [https://www.cff.org/medical-professionals/patient-registry]
- Ramsey BW, Davies J, McElvaney NG, Tullis E, Bell SC, Drevinek P, Griese M, McKone EF, Wainwright CE, Konstan MW et al. A CFTR potentiator in patients with cystic fibrosis and the G551D mutation. N Engl J Med. 2011; 365(18):1663-1672.
- Wainwright CE, Elborn JS, Ramsey BW, Marigowda G, Huang X, Cipolli M, Colombo C, Davies JC, De Boeck K, Flume PA et al: Lumacaftor-Ivacaftor in Patients with Cystic Fibrosis Homozygous for Phe508del CFTR. N Engl J Med. 2015; 373(3):220-231.
- Taylor-Cousar JL, Munck A, McKone EF, van der Ent CK, Moeller A, Simard C, Wang LT, Ingenito EP, McKee C, Lu Y et al. Tezacaftor-Ivacaftor in Patients with Cystic Fibrosis Homozygous for Phe508del. N Engl J Med. 2017; 377(21):2013-2023.
- Rowe SM, Daines C, Ringshausen FC, Kerem E, Wilson J, Tullis E, Nair N, Simard C, Han L, Ingenito EP et al. Tezacaftor-Ivacaftor in Residual-Function Heterozygotes with Cystic Fibrosis. N Engl J Med. 2017; 377(21):2024-2035.
- Middleton PG, Mall MA, Drevinek P, Lands LC, McKone EF, Polineni D, Ramsey BW, Taylor-Cousar JL, Tullis E, Vermeulen F et al. Elexacaftor-Tezacaftor-Ivacaftor for Cystic Fibrosis with a Single Phe508del Allele. N Engl J Med. 2019; 381(19):1809-1819.
- Heijerman HGM, McKone EF, Downey DG, Van Braeckel E, Rowe SM, Tullis E, Mall MA, Welter JJ, Ramsey BW, McKee CM et al. Efficacy and safety of the elexacaftor plus tezacaftor plus ivacaftor combination regimen in people with cystic fibrosis homozygous for the F508del mutation: a double-blind, randomised, phase 3 trial. Lancet. 2019; 394(10212):1940-1948.
- Barry PJ, Mall MA, Alvarez A, Colombo C, de Winter-de Groot KM, Fajac I, McBennett KA, McKone EF, Ramsey BW, Sutharsan S et al. Triple Therapy for Cystic Fibrosis Phe508del-Gating and -Residual Function Genotypes. N Engl J Med. 2021; 385(9):815-825.
- Bessonova L, Volkova N, Higgins M, Bengtsson L, Tian S, Simard C, Konstan MW, Sawicki GS, Sewall A, Nyangoma S et al. Data from the US and UK cystic fibrosis registries support disease modification by CFTR modulation with ivacaftor. Thorax. 2018; 73(8):731-740.
- Burgel PR, Durieu I, Chiron R, Ramel S, Danner-Boucher I, Prevotat A, Grenet D, Marguet C, Reynaud-Gaubert M, Macey J et al. Rapid Improvement after Starting Elexacaftor-Tezacaftor-Ivacaftor in Patients with Cystic Fibrosis and Advanced Pulmonary Disease. Am J Respir Crit Care Med. 2021; 204(1):64-73.
La myofibromatose infantile : de l’analyse génétique au traitement
Jean-Baptiste Demoulin
Secteur des Sciences de la Santé, Faculté de Pharmacie et des Sciences Biomédicales|FASB De Duve Institute|DDUV,B-1200 Brussels, Belgium
Le myofibrome est une tumeur périvasculaire bénigne présentant une localisation cutanée, musculaire ou osseuse. Il peut régresser spontanément ou nécessiter une intervention chirurgicale mineure. La présence de multiples myofibromes, généralement observée chez de très jeunes enfants, définit la myofibromatose infantile, une maladie rare potentiellement mortelle (1). Ces tumeurs peuvent en effet perturber sévèrement le bon fonctionnement des organes internes par compression ou occlusion. Une chimiothérapie (vinblastine et méthotrexate) est alors nécessaire.
Dans le cadre d’une collaboration entre l’institut de Duve et le service d’hématologie et oncologie pédiatrique des Cliniques universitaires Saint-Luc, nous avons identifié des mutations du gène PDGFRB dans plus de 80% des formes sévères de la maladie (2). Dans la majorité des cas, ces altérations génétiques sont restreintes à la tumeur, mais une forme familiale a également été décrite. Etonnamment, de nombreux patients présentent deux mutations successives dans le même allèle du gène PDGFRB. Ces modifications étant spécifiques des myofibromes, elles présentent un grand intérêt pour faciliter le diagnostic de la maladie.
Le gène PDGFRB (platelet-derived growth factor receptor beta) produit un récepteur à activité tyrosine kinase. Notre laboratoire a démontré que les mutations observées dans les myofibromes activent ce récepteur de façon aberrante, conduisant à une prolifération incontrôlée des myofibroblastes. In vitro, cet effet peut être bloqué par l’imatinib (Glivec), un inhibiteur de tyrosine kinases utilisé depuis vingt ans pour traiter la leucémie myéloïde chronique. Sur base de ces observations, ce médicament a été administré à une dizaine de patients dans le monde avec des résultats très prometteurs, qui devront être confirmés par un essai clinique (3). Le principal effet secondaire de l’imatinib chez l’enfant est un ralentissement réversible de la croissance.
Dans des cas exceptionnels, certains variants du gène PDGFRB ont été associés à d’autres pathologies (Figure 1). Il s’agit notamment d’anévrismes fusiformes, du syndrome de Penttinen et du syndrome de surcroissance de Kosaki. Dans ce dernier cas, les mutations de PDGFRB sont germinales et peuvent être associées à la présence de myofibromes. D’autres variants conduisant à une perte de fonction du récepteur sont responsables de calcifications familiales primaires du cerveau, associées à des troubles neurologiques (4). Nos travaux ont permis de mieux comprendre comment le même gène pouvait être lié à ces différentes pathologies.
En conclusion, cette étude a démontré que des mutations du récepteur PDGFRB sont à l’origine de la majorité des cas de myofibromatose infantile, ouvrant la voie à un nouveau test diagnostique et à un nouveau traitement.

Références
- Hettmer, S., Dachy, G., Seitz, G., Agaimy, A., Duncan, C., Jongmans, M., Hirsch, S., Kventsel, I., Kordes, U., de Krijger, R. R., Metzler, M., Michaeli, O., Nemes, K., Poluha, A., Ripperger, T., Russo, A., Smetsers, S., Sparber-Sauer, M., Stutz, E., Bourdeaut, F., Kratz, C. P., and Demoulin, J. B. (2020) Genetic testing and surveillance in infantile myofibromatosis: a report from the SIOPE Host Genome Working Group. Fam Cancer
- Dachy, G., De Krijger, R. R., Fraitag, S., Theate, I., Brichard, B., Hoffman, S. B., Libbrecht, L., Arts, F. A., Brouillard, P., Vikkula, M., Limaye, N., and Demoulin, J. B. (2019) Association of PDGFRB mutations with pediatric myofibroma and myofibromatosis. JAMA dermatology 155, 946-950
- Pond, D., Arts, F. A., Mendelsohn, N. J., Demoulin, J. B., Scharer, G., and Messinger, Y. (2018) A Patient with Germline Gain-of-Function PDGFRB N666H Mutation and Marked Clinical Response to Imatinib. Genetics in Medicine 20, 142-150
- Lenglez, S., Sablon, A., Fenelon, G., Boland, A., Deleuze, J. F., Boutoleau-Bretonniere, C., Nicolas, G., and Demoulin, J. B. (2022) Distinct functional classes of PDGFRB pathogenic variants in primary familial brain calcification. Hum Mol Genet 31, 399-409