Introduction
Les néoplasies myéloprolifératives (MPN) classiques BCR-ABL1 négatives se développent suite à une prolifération clonale d’une seule cellule souche hématopoïétique qui sera responsable d’une production excessive de cellules myéloïdes matures. Les 3 sous-types les plus fréquents sont la maladie de Vaquez (polycythemia vera, PV), la thrombocytémie essentielle (TE) et la myélofibrose primitive (MF) ou post- PV/post-TE. L’acquisition d’une des 3 mutations driver (JAK2V617F, Calréticuline (CALR), myeloproliferative leukemia protein (MPL)/thrombopoiétin receptor (TPOR)) va activer constitutivement la voie de signalisation JAK/STAT dans la cellule et initier et promouvoir le développement du MPN. L’activation pathologique de JAK/STAT est également décrite chez les patients triple négatifs (+- 5-10% des patients) probablement par d’autres mutations driver à découvrir.
La clinique des MPN
Les symptômes
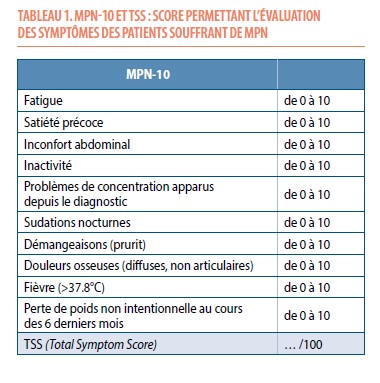
Ces néoplasies myéloprolifératives sont des maladies chroniques. Les symptômes dépendent de chaque sous-type. Cependant, des symptômes communs à ces 3 entités peuvent interférer avec la vie sociale, la vie professionnelle, l’activité physique, l’indépendance aux tâches journalières et la qualité de vie (1). Ces symptômes sont parfois difficiles à relier à la pathologie pour le médecin. Le MPN-10 permet d’évaluer l’intensité des 10 symptômes les plus fréquents dans les MPN (Tableau 1). Le TSS (Total Symptom Score) est la somme des 10 résultats (2). Ce score est utilisé par l’hématologue en consultation pour le suivi du patient et également dans les études cliniques pour évaluer l’efficacité de nouvelles thérapies.


La maladie de Vaquez (PV) peut se manifester par des troubles microvasculaires (céphalées, vertiges, troubles visuels de type vision floue ou scotomes, palpitations, douleur thoracique, érythromélalgies et paresthésies des extrémités), du prurit (48%), de l’inconfort abdominal lié à la splénomégalie, des symptômes constitutionnels (fièvre, sudations nocturnes) (14%), des thromboses (25%) ou des hémorragies (4%) (3). Les érythromélalgies sont caractérisées par un érythème, une chaleur et des douleurs aux extrémités distales. Elles sont probablement liées à des interactions anormales entre les plaquettes et l’endothélium (4). Le prurit ‘aquagénique’ favorisé par l’eau chaude est fréquemment décrit par les patients.
La thrombocytémie essentielle (TE) peut être asymptomatique ou se manifester par des céphalées, des troubles microcirculatoires comme des érythromélalgies, des symptômes constitutionnels ou des complications thrombotiques ou hémorragiques (5). La splénomégalie est rare ou de taille limitée (6).
La myélofibrose primitive (MF) se manifeste par une anémie sévère, une hépato-splénomégalie importante, des symptômes constitutionnels, de la cachexie, des douleurs osseuses, des thromboses ou hémorragies (7).
Le diagnostic
Les critères diagnostiques révisés récemment (classifications ICC-2022, WHO-2022) insistent sur la nécessité d’intégrer les comptages des cellules sanguines, les données moléculaires et l’analyse morphologique de la biopsie médullaire. Aucun de ces 3 paramètres ne peut seul suffire au diagnostic (8, 9).
Le diagnostic de la maladie de Vaquez (PV) se base sur les critères de la classification ICC-2022 (8) et requiert les 3 critères majeurs ou les 2 premiers critères majeurs et le critère mineur (Tableau 2).

La biopsie médullaire permet d’évaluer un degré de fibrose initial (présent chez 20% des patients); ce qui prédit une progression plus rapide en myélofibrose post-PV chez ces patients. La ponction médullaire permet également d’obtenir un caryotype dont certaines altérations sont associées à une survie inférieure dans la PV. L’étude des mutations par Next Génération Sequencing (NGS) bien qu’ayant un impact pronostique n’est actuellement pas remboursée en Belgique. Une détection de la mutation JAK2V617F avec une fréquence allélique < 1% nécessite de réaliser une ponction-biopsie médullaire.
Le diagnostic de la thrombocytémie essentielle (TE) se base sur les critères de la classification ICC-2022 (8) et requiert tous les critères majeurs ou les 3 premiers critères majeurs associés à un critère mineur (Tableau 3). Pour les 10-15% de patients sans aucune des 3 mutations drivers détectée, la biopsie médullaire va permettre d’établir le diagnostic de TE triple négative.

La biopsie médullaire est primordiale pour distinguer la TE de la forme pré-fibrotique avec thrombocytose de la myélofibrose primitive. La morphologie des mégacaryocytes permet d’en faire la distinction. Dans la myélofibrose primitive, les mégacaryocytes présentent un haut degré d’atypies : ils sont de taille variable (petits à géants) avec des défauts fréquents de maturation et regroupés en clusters denses. Le caryotype et le NGS permettront d’établir le pronostic de la myélofibrose pré-fibrotique et de définir le traitement. Le risque d’évolution en myélofibrose établie est dans la forme pré-fibrotique de 10% à 10 ans versus < 5% dans le TE. Le pronostic des 2 entités est très différent.
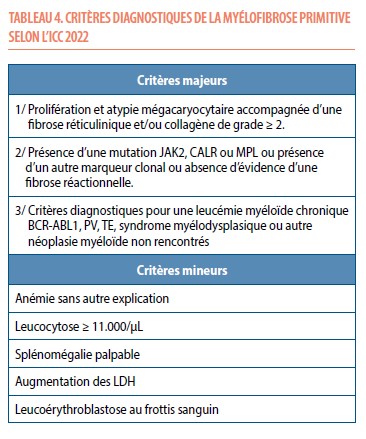
Le diagnostic de la myélofibrose primitive repose également sur les critères de la classification ICC-2022 (7) et requiert les 3 critères majeurs et 1 critère mineur (Tableau 4).
Le diagnostic de myélofibrose post-PV ou post-TE se base sur les critères de l’IWG-MRT qui ne seront pas détaillés dans cette revue.
Le pronostic
La survie médiane des patients atteints de maladie de Vaquez est rapportée à 12.7 ans sur une série de patients traités à la Mayo Clinic entre 2000 et 2017 (10). La survie dépend de l’âge : ≤ 40 ans : 37 ans, 41-60 ans : 22 ans et > 60 ans : 10 ans. Les autres facteurs pronostiques sont la leucocytose, les thromboses, les anomalies chromosomiques et les mutations SRSF2, IDH2, RUNX1, U2AF1 (11). Les risques thrombotiques sont de 26% à 20 ans (12). Le risque d’évolution en myélofibrose post-PV est de 6-14% à 15 ans (13). Le risque de transformation en une phase blastique (leucémie myéloïde aiguë) est de 2.3% à 10 ans et 5.5% à 15 ans (14).
La survie médiane de patients atteints de thrombocytémie essentielle est de 14.9 ans (10). La survie médiane dépend également de l’âge : < 60 ans : 32.7 ans (15). Les autres facteurs de risque influençant la survie sont la leucocytose, l’historique thrombotique et les mutations SF3B1 et SRSF2 (11). Le risque cumulatif d’une évolution en myélofibrose post-ET est de 4-11% à 15 ans (13). Les mutations U2AF1 et SF3B1 affectent le risque d’évolution en myélofibrose. Le risque de transformation en une phase blastique est de 5% à 20 ans.
La survie médiane de patients atteints de myélofibrose primitive est de 4.4 ans (10). Le risque de transformation en leucémie aigüe est de 6 à 21% à 5 ans (16). La survie est influencée par l’âge, l’anémie, la leucocytose, la blastose, la thrombopénie, les symptômes constitutionnels, les anomalies chromosomiques et les mutations ASXL1, SRSF2, EZH2, IDH1, IDH2, U2AF1.
Le traitement en pratique clinique en 2023 en résumé
Le traitement actuel de la maladie de Vaquez a pour but d’éviter les thromboses (3). Il requiert pour tous les patients la prise d’Asaflow 80 mg 1x/jour et des saignées pour maintenir un taux d’hématocrite < 45% (3) (Figure 1).
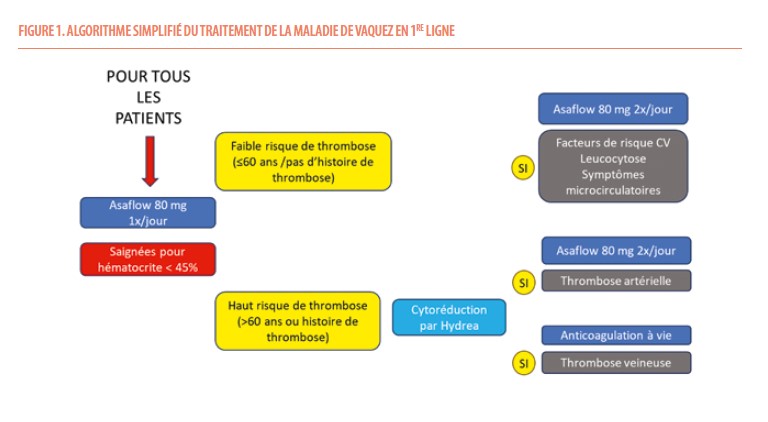
Les patients sont classés en faible risque thrombotique (< 60 ans, pas d’histoire de thrombose) et haut risque thrombotique (> 60 ans OU histoire de thrombose).
*Il est recommandé de doubler l’Asaflow chez des patients à faible risque thrombotique avec des facteurs de risque cardiovasculaires ou une leucocytose ou des symptômes microcirculatoires. L’Asaflow prise 2x/jour est plus efficace pour réduire l’activation plaquettaire mesurée par le taux sérique de thromboxane B2 (17).
*Les patients à haut risque thrombotique seront cytoréduits par hydroxycarbamide (Hydrea®) en 1re ligne. Attention que l’Hydrea est tératogène. Si le patient a un antécédent de thrombose artérielle, l’Asaflow sera prescrit 2x/jour. Si le patient a un antécédent de thrombose veineuse, le patient sera anticoagulé à vie.
La leucocytose doit être contrôlée idéalement < 11.000/µL (au minimum < 15.000/µL) de même que les facteurs de risque cardiovasculaires. Le ruxolitinib (Jakavi®) est remboursé chez les patients résistants ou intolérants à l’Hydrea.
Le traitement actuel de la thrombocytémie essentielle a également pour but d’éviter les thromboses et les hémorragies en cas de thrombocytose extrême (5). Les patients sont classés selon le score IPSET révisé à très faible ou faible risque thrombotique (± JAK2V617F, < 60 ans, pas d’histoire de thrombose) et risque thrombotique intermédiaire ou haut (± JAK2V617F, > 60 ans OU histoire de thrombose) (Figure 2) (18). Les patients avec un taux de plaquettes > 1.500.000/µL seront cytoréduits par hydroxycarbamide (Hydrea®) pour diminuer le risque hémorragique. La maladie de von Willebrand acquise asymptomatique n’est pas une indication de cytoréduction sauf avant chirurgie.
*Les patients classés à très faible risque thrombotique (ABSENCE de mutation JAK2V617F, ≤ 60 ans, PAS d’histoire de thrombose) en l’absence de facteur de risque cardiovasculaire et en l’absence de mutation driver identifiée (triple négatifs) seront suivis. En présence de facteur de risque cardiovasculaire ou de la mutation CALR/MPL, ces patients seront traités par Asaflow 80 mg 1x/jour. L’utilisation de l’aspirine sera évitée chez les patients avec une thrombocytose extrême > 1.000.000/µL et une maladie de von Willebrand acquise (activité cofacteur ristocétine < 20%).
*Les patients classés à faible risque thrombotique (mutation JAK2V617F, ≤ 60 ans, pas d’histoire de thrombose) en l’absence de facteur de risque cardiovasculaire seront traités par Asaflow 80 mg 1x/jour. L’Asaflow sera doublée soit 2x/jour chez ces patients avec facteur de risque cardiovasculaire.
*Les patients classés à risque thrombotique intermédiaire (ABSENCE de mutation JAK2V617F, > 60 ans, pas d’histoire de thrombose) seront traités par Asaflow 1x/jour et le plus souvent cytoréduits par hydroxycarbamide (Hydrea®). Le taux de plaquettes visé est < 400.000/µL soit une normalisation.
*Les patients classés à haut risque thrombotique (> 60 ans ou histoire de thrombose) seront traités par Asaflow 80 mg 1x/jour et cytoréduits par hydroxycarbamide (Hydrea®).
Si le patient a un antécédent de thrombose artérielle, l’Asaflow sera prescrit ٢x/jour. Si le patient a un antécédent de thrombose veineuse, le patient sera anticoagulé à vie. Chez ces patients JAK2V617F positifs, on considère dans certaines situations d’ajouter l’Asaflow tout en pesant les risques hémorragiques.
La leucocytose doit être contrôlée idéalement < 11.000/µL (au minimum < 15.000/µL) de même que les facteurs de risque cardiovasculaires.

Le traitement actuel de la myélofibrose est basé sur des scores pronostiques qui incluent les paramètres cliniques, biologiques, les anomalies cytogénétiques et moléculaires. Les patients avec un score intermédiaire -2 ou élevé seront orientés vers une allogreffe de cellules souches périphériques à visée curative. La splénomégalie sera réduite préalablement par inhibiteur de JAK2 de type ruxolitinib (Jakavi ®) ou par splénectomie en cas d’échec. Les patients à risque faible ou intermédiaire -1 seront suivis régulièrement. Les patients à risque intermédiaire -1 ou plus ayant une splénomégalie et/ou des symptômes (listés dans le score MPN10) seront traités par Jakavi. Ce traitement oral permet une diminution des symptômes de ≥ 50% chez 46% des patients et une diminution de la splénomégalie de ≥ 35% chez 42% des patients (20). Le Jakavi améliore la qualité de vie des patients et possiblement leur survie. Le Jakavi peut transitoirement aggraver l’anémie et nécessiter des transfusions de globules rouges pendant les 12 premières semaines de traitement. L’efficacité du Jakavi est malheureusement transitoire soit 3-5 ans. Le phénomène de persistance de la cellule muté a été décrit. Pour les patients résistants au Jakavi, un traitement par un autre inhibiteur de JAK2 - fedratinib (Inrebic®) est actuellement disponible et permet une diminution des symptômes de ≥ 50% chez 31% des patients et une diminution de la splénomégalie de ≥ 35% chez 27% des patients (20). D’autres inhibiteurs de JAK2 (momelotinib, pacritinib) ont montré leur efficacité et seront bientôt accessibles pour certains patients notamment avec anémie ou thrombopénie. De rares patients seront cytoréduits par Hydrea s’ils présentent une forme proliférative débutante.
L’espoir d’un futur traitement curatif des MPN …
Tous ces traitements excepté l’allogreffe dans la myélofibrose ne sont pas curatifs. Ils permettent dans la maladie de Vaquez et dans la thrombocytémie essentielle de diminuer les risques de thrombose et d’hémorragie mais n’empêchent pas une évolution en myélofibrose ou en leucémie aigüe. Seul l’interferon (Pegasys®, Besremi®), malheureusement non remboursé en Belgique dans les MPN, permet d’obtenir une diminution du clone muté voire une réponse moléculaire complète (JAK2V617F indétectable) chez 14% de patients traités par Pegasys pour une maladie de Vaquez et 30% par Besremi (21, 22). La réduction du clone a été associée à une diminution du risque d’évolution (23). Dans la myélofibrose, les patients développent des résistances aux inhibiteurs de JAK2.
Les progrès récents de la recherche sont encourageants notamment pour cibler les cellules avec la mutation de la calréticuline. Des travaux de recherche réalisés récemment à l’Institut de Duve sur la compréhension de la structure de la calréticuline mutée, de ses interactions avec le récepteur à la thrombopoïtéine (TpoR) et de la structure de ce récepteur TpoR ont et vont permettre des développements thérapeutiques ultérieurs (24). L’identification précise des résidus du TpoR impliqués dans l’activation spécifique par JAK2V617F pourrait également ouvrir la porte à de nouvelles thérapies modulant la conformation du TpoR (25).
Lors du dernier congrès de la société américaine d’hématologie, Reis et al ont montré l’efficacité d’un anticorps monoclonal humain IgG1 mAb INCA033989 ciblant uniquement la calréticuline mutée dans des lignées cellulaires murines, des cellules murines in vitro et des cellules CD34+ dérivées de patients. Cet anticorps inhibe l’activation du TpoR par la calréticuline mutée (26). Des souris transplantées ont été traitées avec cet anticorps. Après 10 semaines de traitement, on n’observe pas de thrombocytose mais une diminution significative du nombre de cellules progénitrices calréticuline mutées ainsi que des mégacaryocytes dans la moelle sans affecter les cellules non mutées. Les résultats de l’étude clinique de phase 1 chez des patients souffrant de MPN avec la mutation de la calréticuline devraient être présentés prochainement.
Enfin, un anticorp bi-specifique (JNJ-88549968) produit par Janssen Johnson & Johnson en collaboration avec MyeloPro GmbH (Vienne) et des chercheurs de l’Institut de Duve cible la calréticuline mutée à la surface du clone MPN et la protéine CD3 des lymphocytes T. L’effet cancéreux de la calréticuline mutée requiert sa présence à la surface cellulaire (27). Ainsi les lymphocytes T recrutés contre les cellules qui portent la mutation calréticuline à leur surface vont éliminer in vitro sur du sang total les cellules progénitrices et CD34+ du clone MPN car elles sont les seules cellules qui portent la mutation de la calréticuline (28). Ce traitement sera testé en clinique car il a le potentiel d’éradiquer la maladie par l’élimination des cellules souches du clone MPN sans atteindre les cellules non mutées.
En conclusion
Les néoplasies myéloprolifératives sont actuellement des maladies chroniques responsables de symptômes altérant la qualité de vie des patients ou de thromboses associées à des séquelles neurologiques, respiratoires, hépatiques ou autres. Certains patients évoluent vers une myélofibrose ou une leucémie de pronostic très réservé. Les avancées récentes dans la recherche ont permis une meilleure compréhension du mécanisme physiopathologique de ces maladies et le développement de futures nouvelles approches thérapeutiques à visée curative.
Affiliations
1. Service d’hématologie, Cliniques universitaires Saint-Luc, Bruxelles, Belgique.
2. Université catholique de Louvain and de Duve Institute, Bruxelles, Belgique.
3. Ludwig Institute for Cancer Research Brussels, Bruxelles, Belgique.
4. Wel Research Institute, WelBio Department, Havren, Belgique.
5. Ludwig Institute for Cancer Research, Nuffield Department of Medicine, Oxford University, Oxford, UK.
Correspondance
Dre Violaine Havelange
Université catholique de Louvain and de Duve Institute
Cliniques universitaires Saint-Luc
Service d’hématologie
Avenue Hippocrate 10
B-1200 Bruxelle
Références
- Mesa R, Miller CB, Thyne M, Mangan J, Goldberger S, Fazal S, et al. Myeloproliferative neoplasms (MPNs) have a significant impact on patients’ overall health and productivity: the MPN Landmark survey. BMC Cancer. 2016;16:167.
- Emanuel RM, Dueck AC, Geyer HL, Kiladjian JJ, Slot S, Zweegman S, et al. Myeloproliferative neoplasm (MPN) symptom assessment form total symptom score: prospective international assessment of an abbreviated symptom burden scoring system among patients with MPNs. J Clin Oncol. 2012;30(33):4098-103.
- Tefferi A, Barbui T. Polycythemia vera: 2024 update on diagnosis, risk-stratification, and management. Am J Hematol. 2023;98(9):1465-87.
- Michiels JJ, Abels J, Steketee J, van Vliet HH, Vuzevski VD. Erythromelalgia caused by platelet-mediated arteriolar inflammation and thrombosis in thrombocythemia. Ann Intern Med. 1985;102(4):466-71.
- Tefferi A, Vannucchi AM, Barbui T. Essential thrombocythemia: 2024 update on diagnosis, risk stratification, and management. Am J Hematol. 2024;99(4):697-718.
- Godfrey AL, Green AC, Harrison CN. Essential thrombocythemia: challenges in clinical practice and future prospects. Blood. 2023;141(16):1943-53.
- Tefferi A. Primary myelofibrosis: 2023 update on diagnosis, risk-stratification, and management. Am J Hematol. 2023;98(5):801-21.
- Arber DA, Orazi A, Hasserjian RP, Borowitz MJ, Calvo KR, Kvasnicka HM, et al. International Consensus Classification of Myeloid Neoplasms and Acute Leukemias: integrating morphologic, clinical, and genomic data. Blood. 2022;140(11):1200-28.
- Khoury JD, Solary E, Abla O, Akkari Y, Alaggio R, Apperley JF, et al. The 5th edition of the World Health Organization Classification of Haematolymphoid Tumours: Myeloid and Histiocytic/Dendritic Neoplasms. Leukemia. 2022;36(7):1703-19.
- Smith CJ, Thomas JW, Ruan G, Hyun MC, Bansal R, McLaughlin N, et al. A population-based study of outcomes in polycythemia vera, essential thrombocythemia, and primary myelofibrosis in the United States from 2001 to 2015: Comparison with data from a Mayo Clinic single institutional series. Am J Hematol. 2021;96(12):E464-E8.
- Tefferi A, Guglielmelli P, Lasho TL, Coltro G, Finke CM, Loscocco GG, et al. Mutation-enhanced international prognostic systems for essential thrombocythaemia and polycythaemia vera. Br J Haematol. 2020;189(2):291-302.
- Szuber N, Mudireddy M, Nicolosi M, Penna D, Vallapureddy RR, Lasho TL, et al. 3023 Mayo Clinic Patients With Myeloproliferative Neoplasms: Risk-Stratified Comparison of Survival and Outcomes Data Among Disease Subgroups. Mayo Clin Proc. 2019;94(4):599-610.
- Cerquozzi S, Tefferi A. Blast transformation and fibrotic progression in polycythemia vera and essential thrombocythemia: a literature review of incidence and risk factors. Blood Cancer J. 2015;5(11):e366.
- Tefferi A, Rumi E, Finazzi G, Gisslinger H, Vannucchi AM, Rodeghiero F, et al. Survival and prognosis among 1545 patients with contemporary polycythemia vera: an international study. Leukemia. 2013;27(9):1874-81.
- Tefferi A, Guglielmelli P, Larson DR, Finke C, Wassie EA, Pieri L, et al. Long-term survival and blast transformation in molecularly annotated essential thrombocythemia, polycythemia vera, and myelofibrosis. Blood. 2014;124(16):2507-13; quiz 615.
- Tefferi A, Lasho TL, Jimma T, Finke CM, Gangat N, Vaidya R, et al. One thousand patients with primary myelofibrosis: the mayo clinic experience. Mayo Clin Proc. 2012;87(1):25-33.
- De Stefano V, Rocca B, Tosetto A, Soldati D, Petrucci G, Beggiato E, et al. The Aspirin Regimens in Essential Thrombocythemia (ARES) phase II randomized trial design: Implementation of the serum thromboxane B(2) assay as an evaluation tool of different aspirin dosing regimens in the clinical setting. Blood Cancer J. 2018;8(6):49.
- Barbui T, Vannucchi AM, Buxhofer-Ausch V, De Stefano V, Betti S, Rambaldi A, et al. Practice-relevant revision of IPSET-thrombosis based on 1019 patients with WHO-defined essential thrombocythemia. Blood Cancer J. 2015;5(11):e369.
- Verstovsek S, Mesa RA, Gotlib J, Levy RS, Gupta V, DiPersio JF, et al. A double-blind, placebo-controlled trial of ruxolitinib for myelofibrosis. N Engl J Med. 2012;366(9):799-807
- Harrison CN, Schaap N, Vannucchi AM, Kiladjian JJ, Tiu RV, Zachee P, et al. Janus kinase-2 inhibitor fedratinib in patients with myelofibrosis previously treated with ruxolitinib (JAKARTA-2): a single-arm, open-label, non-randomised, phase 2, multicentre study. Lancet Haematol. 2017;4(7):e317-e24.
- Quintas-Cardama A, Kantarjian H, Manshouri T, Luthra R, Estrov Z, Pierce S, et al. Pegylated interferon alfa-2a yields high rates of hematologic and molecular response in patients with advanced essential thrombocythemia and polycythemia vera. J Clin Oncol. 2009;27(32):5418-24.
- Kiladjian JJ, Klade C, Georgiev P, Krochmalczyk D, Gercheva-Kyuchukova L, Egyed M, et al. Long-term outcomes of polycythemia vera patients treated with ropeginterferon Alfa-2b. Leukemia. 2022;36(5):1408-11.
- Abu-Zeinah G, Krichevsky S, Cruz T, Hoberman G, Jaber D, Savage N, et al. Interferon-alpha for treating polycythemia vera yields improved myelofibrosis-free and overall survival. Leukemia. 2021;35(9):2592-601.
- Papadopoulos N, Nedelec A, Derenne A, Sulea TA, Pecquet C, Chachoua I, et al. Oncogenic CALR mutant C-terminus mediates dual binding to the thrombopoietin receptor triggering complex dimerization and activation. Nat Commun. 2023;14(1):1881.
- Papadopoulos N, Pristavec A, Nedelec A, Levy G, Staerk J, Constantinescu SN. Modulation of Human Thrombopoietin Receptor Conformations Uncouples JAK2 V617F-Driven From Cytokine-Induced Activation. Blood. 2023.
- Reis E, Buonpane R, Celik H, Marty C, Lei A, Jobe F, et al. Discovery of INCA033989, a monoclonal antibody that selectively antagonizes mutant Calreticulin oncogenic function in myeloproliferative neoplasms (MPNs). Blood. 2022;140:14-5: ASH Abstract.
- Pecquet C, Papadopoulos N, Balligand T, Chachoua I, Tisserand A, Vertenoeil G, et al. Secreted mutant calreticulins as rogue cytokines in myeloproliferative neoplasms. Blood. 2023;141(8):917-29.
- Samakai E, Balmaña M, Janssen L, Amorim R, Cornelissen I, Hug E, et al. Novel and disease-modifying therapy to treat myeloproliferative neoplasms: CALRmutxCD3 bispecific antibody. Blood. 2023;In press: ASH abstract.