Introduction
Le syndrome de fuite capillaire idiopathique ou maladie de Clarkson est une pathologie rare, de pronostic sévère et dont la physiopathologie reste mal connue. Caractérisé cliniquement par une prise pondérale transitoire avec œdèmes, oligurie, hypotension artérielle pouvant aller jusqu’à l’état de choc hypovolémique et biologiquement par l’association quasi pathognomonique d’une hémoconcentration et d’une hypoalbuminémie paradoxale sans albuminurie, il survient par crises récidivantes liées à un accès brutal d’hyperperméabilité capillaire laissant passer massivement des protéines plasmatiques dans le secteur extravasculaire, souvent sur épisode infectieux déclencheur. Moins de 250 cas (1) ont été rapportés dans la littérature depuis 1960, mais le syndrome est probablement sous-diagnostiqué au vu de ses signes aspécifiques – aisément confondus avec ceux d’un choc septique, d’une décompensation cardiaque, d’un angio-œdème ou d’un choc anaphylactique – et de sa mortalité élevée, oscillant entre 30 et 76% (2,3,4).
Vignette clinique
À titre illustratif, nous rapportons ici le cas d’un patient de 42 ans chez qui le diagnostic de syndrome de Clarkson a été posé devant la survenue brutale d’un tableau œdémateux avec hypovolémie relative et dissociation entre l’albumine et l’hématocrite dans un contexte de détresse respiratoire aiguë sur pneumopathie infectieuse virale.
Le patient, sans antécédent notable hormis un asthme dans l’enfance et un tabagisme actif, consulte aux urgences pour une dyspnée d’effort associée à l’apparition d’œdèmes des membres et de la face, dans un contexte de rhinopharyngite. En première hypothèse est évoqué un angio-œdème bradykinique, le patient ayant vécu, suite à une rhinopharyngite virale, un épisode similaire spontanément résolutif 1 an auparavant. Un traitement d’épreuve par anti-fibrinolytique (Exacyl®) est alors mis en place, mais se révèle inefficace.
Devant une hémoconcentration (Hb 17.7 g/dL ; Htc 54%) et une hypoalbuminémie paradoxale (26,7 g/L) sur la biologie, il est suspecté un syndrome de Clarkson, et une cure d’immunoglobulines polyvalentes est initiée durant 48h, à raison de 1g/kg/j. Suite à une instabilité hémodynamique (TA 86/55 ; FC 95 bpm) et une détresse respiratoire aiguë, le patient est transféré en réanimation, où l’on recourt alors à l’oxygénothérapie nasale à haut débit (Optiflow®) avec 80% de FiO2, ainsi qu’à une hydratation prudente au sérum salé isotonique. L’examen clinique retrouve des œdèmes des membres et des sibilants diffus avec des crépitants basaux à l’auscultation pulmonaire. L’échographie cardiaque ne met pas de péricardite en évidence, l’électrocardiogramme ne déroule qu’une tachycardie sinusale, la fibroscopie ne détecte pas d’anomalie des cordes vocales, mais la radiographie de thorax objective un syndrome alvéolo-interstitiel bilatéral prédominant en base droite, pour lequel est initiée une antibiothérapie par céphalosporine (Claforan®) et macrolide (Rovamycine®) durant 7 jours. Un bilan infectieux complet est réalisé : les antigénuries légionelle et pneumocoque reviennent négatives mais la PCR revient positive pour la grippe A-H3, permettant de conclure à une pneumopathie virale pour laquelle un traitement par antiviral (Tamiflu®) durant 5 jours est instauré.
L’évolution clinique est favorable avec une régression des œdèmes sans recours aux diurétiques, et un amendement de la dyspnée, avec un relais aux lunettes à 1L/min d’O2. L’évolution des anomalies biologiques est également favorable, mettant toutefois en évidence – outre l’hémoconcentration et l’hypoalbuminémie caractéristiques du syndrome de fuite capillaire idiopathique – un épisode de rhabdomyolyse avec des CPK à 1843 U/L, témoin d’un syndrome des loges secondaire aux œdèmes des membres inférieurs et aggravé par le remplissage. Les douleurs musculaires ainsi que les CPK régressent à l’arrêt de ce dernier.
Par ailleurs, le test de Landis réalisé – test étudiant les courbes de décroissance plasmatique en clinostatisme et en orthostatisme de la Sérum-albumine radio-iodée (I-125) par voie IV – objective une fuite protéique interstitielle anormale, confirmant le diagnostic de syndrome de Clarkson. À noter un pic monoclonal IgG kappa à 5,3 g/L et un rapport de chaines légères kappa/lambda à 1,6, que l’on objectivera un mois plus tard sur l’électrophorèse des protéines, à distance de la cure d’immunoglobulines. Enfin, étant donnée la gravité de la crise avec nécessité de prise en charge en réanimation, il est décidé en réunion de concertation pluridisciplinaire, de la réalisation de cures mensuelles d’immunoglobulines (2g/kg/cure) en prévention de nouvelles crises.
Discussion
Touchant les adultes d’âge moyen de 45ans (5), le syndrome d’hyperperméabilité capillaire reste avant tout un diagnostic clinique, et il convient d’en reconnaître précocement les signes et symptômes. Il peut survenir inopinément ou se manifester par une phase prodromique avec signes aspécifiques généraux (asthénie, malaise, fièvre), digestifs (diarrhées, douleurs abdominales) ou atteinte des voies aériennes supérieures (toux, rhinorrhée), cette dernière étant signalée dans 50% des cas (6). Après la phase prodromique, le syndrome se caractérise par une phase d’état et une phase de récupération. La particularité de la phase d’état est la présence d’une hypotension artérielle sévère voire un choc hypovolémique mais avec conscience préservée, associée à une infiltration œdémateuse des téguments et des séreuses, généralisée ou segmentaire, épargnant initialement le poumon (1,5,6,7). Le profil biologique est celui d’une hémoconcentration majeure avec hypoalbuminémie paradoxale, normalisé en dehors des crises, contrairement à l’électrophorèse des protéines sériques qui révèle dans plus de 80% des cas une gammapathie monoclonale le plus souvent de type immunoglobuline G kappa, pouvant préexister aux épisodes de fuite ou apparaitre durant l’évolution jusqu’à plusieurs années après la première crise (1,2,6). Celle-ci a d’ailleurs fait l’objet de plusieurs études mais son rôle dans la physiopathologie du syndrome de Clarkson reste inconnu, étant donnée sa présence inconstante et sa persistance entre les crises. Cette phase peut se compliquer d’un syndrome des loges avec rhabdomyolyse, dû à l’œdème musculaire et souvent aggravé par un remplissage vasculaire massif, et d’une insuffisance rénale fonctionnelle voire d’une nécrose tubulaire aigue. Il existe également un risque de complications de surcharge aggravées par un remplissage vasculaire massif initial, telles que, outre une rhabdomyolyse, un œdème aigu du poumon et une péricardite. D’autres complications sont encore évoquées dans la littérature, telles qu’une pancréatite, des convulsions, un œdème cérébral, une arythmie cardiaque ou des thromboses (5,7).
La phase de récupération enfin, se caractérise par la normalisation de la tension artérielle, une crise polyurique et la perte pondérale avec disparition des œdèmes par retour de la volémie dans le secteur vasculaire. Devant la récurrence d’un tableau clinico-biologique évocateur, la démarche diagnostique consiste donc prioritairement à exclure les causes secondaires potentielles de fuite capillaire (dont une liste non exhaustive est reprise dans le tableau 1) ou d’œdèmes (Tableau 2), (5,7).
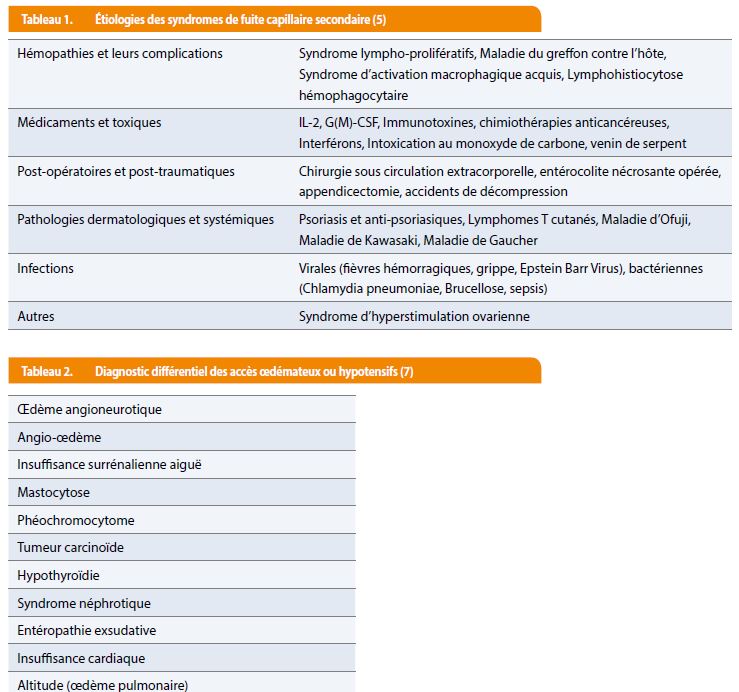
Quelques examens complémentaires sont recommandés dans la littérature en vue d’exclure les plus fréquentes, citons notamment à titre d’exemple : une biologie de routine et des prélèvements bactériologiques pour écarter un sepsis ; la recherche de protéinurie, témoin d’un éventuel syndrome néphrotique ; le dosage et la mesure de l’activité de l’inhibiteur en C1 estérase en faveur d’un œdème angioneurotique ; les taux de NT-proBNP (pro-brain natriuretic peptide) et un bilan complémentaire par électrocardiogramme, échocardiographie et radiographie thoracique afin d’exclure une cause cardiaque, permettant également d’éliminer un syndrome de Takotsubo ou cardiomyopathie de stress qui peut dans certains cas être responsable d’une diminution transitoire de la fraction d’éjection du ventricule gauche suite à une infiltration œdémateuse du myocarde; ou encore un dosage de la tryptase sérique pour exclure une anaphylaxie (1,8). Enfin, le test isotopique de Landis évoqué plus haut – marqueur sensible de la fuite protéique vers le secteur interstitiel – et la détection d’un pic monoclonal sur l’électrophorèse des protéines orientent encore le diagnostic. D’autres tests de réalisation difficile mettant en évidence la fuite d’albumine sont évoqués dans la littérature, tels que le calcul de l’index de Marks – comparant le volume plasmatique évalué à l’aide du volume globulaire et de l’hématocrite, et le volume plasmatique mesuré par albumine marquée – ou des mesures radio-isotopiques de décroissance d’albumine marquée (radioactive iodinated serum albumin ou [RISA]), mais sont considérés comme modérément fiables et de peu d’intérêt en pratique clinique (5,7). Étant donnée la rareté de la maladie, toutes les stratégies thérapeutiques proposées dans la littérature sont basées sur des données observationnelles et non des essais cliniques randomisés ; dès lors le traitement de la phase aiguë reste symptomatique, faisant appel à un remplissage vasculaire prudent, parfois en association avec des amines vasopressives, afin de traiter l’hypovolémie intravasculaire, maintenir la perfusion des organes et éviter une acidose métabolique sévère. Cependant grâce aux avancées des connaissances sur la physiopathologie du syndrome de Clarkson, on sait aujourd’hui qu’il est lié à un réarrangement de la micro-vascularisation par internalisation de protéines jonctionnelles (vascular endothelial cadherins), et à une contraction des cellules endothéliales, médiés durant les crises par une augmentation transitoire de certaines cytokines (Tumor necrosing factor α, Interleukines…) et facteurs angiogéniques tels que VEGF (vascular endothelial growth factor) et Angpt-2 (Angiopoietin 2), (1,4,5). Toutefois, des thérapies ciblées sur ces derniers ont été mises en place dans des cas isolés seulement et les anomalies génétiques responsables, s’il y en a, n’ont pas été identifiées (1,9,10). Outre d’autres traitements symptomatiques et préventifs des complications (Figure 1), il n’existe donc pas à l’heure actuelle de traitement curatif.
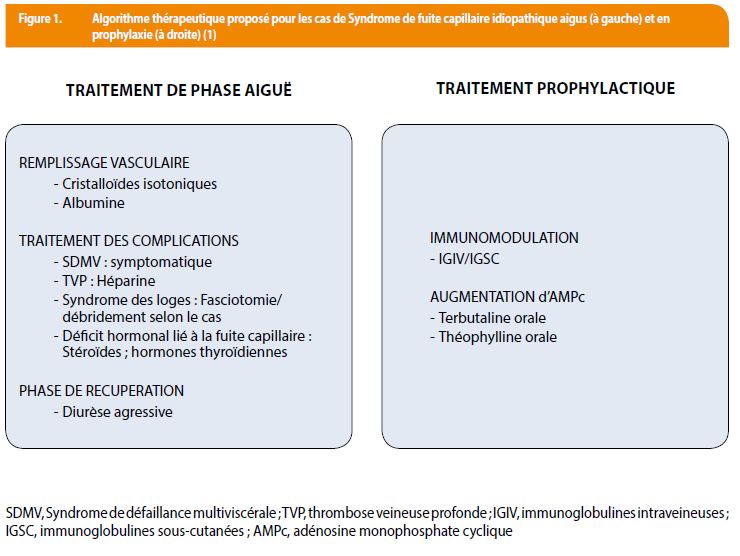
La mise en place d’une prophylaxie au long cours semble être utile, réduisant la mortalité à 5 ans de 80% à 15% (11). Un traitement historique combiné associant terbutaline, un béta-2-mimétique, et théophylline, un inhibiteur de phosphodiestérase, a d’abord été proposé car préviendrait la contraction des cellules endothéliales par augmentation de l’AMPc (adénosine monophosphate cyclique) et inhiberait ainsi l’hyperperméabilité capillaire par stabilisation des cadhérines de la barrière endothéliale. Cependant de janvier 1997 à mars 2016, un registre européen nommé EurêClark, a recensé près de 85% de rechutes malgré ce traitement en prophylaxie ; d’autre part, la tolérance était médiocre avec de nombreux effets indésirables, tels que tremblements, irritabilité, palpitations et insomnies (11,12). Cette thérapie a donc progressivement laissé place aux immunoglobulines à dose immunomodulatrice, proposé en deuxième ligne de traitement prophylactique dès l’an 2000, sous lesquelles 25% de rechutes ont été recensées et depuis lesquelles la sévérité des crises est moindre et leur fréquence moyenne annuelle retombée à 0/patient versus 2,25/patient pour les patients traités par terbutaline et théophylline (1,11,13,14). Il ressort toutefois d’une analyse multivariée récente, publiée en octobre 2017 dans The New American Journal of Medicine et basée sur la plus large cohorte de patients recensée à ce jour (69 patients inclus, sex ratio 1, âge moyen 52 +/- 12 ans), que seules les IgIV et la terbutaline auraient un impact significatif sur la mortalité à long terme et que cette dernière ne diminuerait que la sévérité des crises et non leur fréquence (12). Cette étude a permis d’être plus affirmatif sur l’efficacité des IgIV, recommandant dès lors leur utilisation en première ligne de traitement prophylactique. La tolérance des IgIV est satisfaisante et les rares effets secondaires rapportés – nausées, céphalées, thromboses veineuses et réactions cutanées allergiques non graves – cèdent à l’étalement des perfusions sur quelques jours. Jouissant de propriétés anti-cytokiniques et d’un pouvoir immunomodulateur, leur efficacité en phase aiguë reste toutefois incertaine devant les données limitées de la littérature (1,3,8,14). Le décès (médiane à 47 ans) survient dans 80% des cas en contexte de crise ou de complications liées à la réanimation – notamment œdème aigu du poumon après remplissage vasculaire massif, infections liées à la réanimation, arythmies sous amines vasopressives (5,7). Quelques cas de myélomes ont été également rapportés dans la littérature (5% d’incidence globale après exclusion des formes chroniques), justifiant une surveillance attentive et prolongée des patients (5). Il a d’ailleurs été observé qu’un taux plus élevé de composant monoclonal détecté lors du diagnostic prédisposait à la récurrence de crises plus sévères, incitant à la recherche de son rôle encore mal connu dans la physiopathologie de la maladie (12).
En conclusion, le syndrome d’hyperperméabilité capillaire idiopathique est un syndrome clinico-biologique rare, survenant inopinément ou déclenché par un trigger infectieux, caractérisé par une prise pondérale avec œdème et hypotension artérielle, et par l’association quasi pathognomonique d’une hémoconcentration avec hypoalbuminémie paradoxale sans albuminurie. Même si la compréhension physiopathologique de la maladie fait encore défaut, les études récentes ont permis d’être plus affirmatif quant à l’efficacité des immunoglobulines intraveineuses en première ligne de traitement préventif de nouvelles crises chez les patients pour qui le diagnostic de syndrome de Clarkson avec gammapathie monoclonale a été posé (12).
Recommandations pratiques
1. Le diagnostic doit être évoqué en l’absence d’étiologie évidente ou de cause secondaire de fuite capillaire – et ce d’autant plus si l’on note une gammapathie monoclonale, un caractère récidivant des crises, l’absence d’atteinte pulmonaire initiale et le respect de la conscience malgré une hypotension artérielle effondrée – et devrait conduire à une prise en charge symptomatique précoce en milieu spécialisé.
2. Devant le risque de récidive des crises, les formes graves de la maladie justifient l’instauration d’un traitement d’entretien au long cours, à titre de prophylaxie, où une dose d’IgIV de 2g/kg/mois en 48h est recommandée en première intention.
3. Outre des soins dentaires réguliers, rappelons l’importance d’une vaccination antigrippale et anti-pneumococcique, les crises pouvant être déclenchées par un épisode infectieux.
4. Enfin, au vu de l’importante mortalité, outre une évolution potentielle vers le myélome, un suivi étroit et prolongé des patients doit être assuré afin d’éviter une prise en charge réanimatoire et ses complications.
Affiliations
1 Faculté de médecine et médecine dentaire, Université catholique de Louvain, 1200 Bruxelles, Belgique 2 Service de Médecine interne, Centre Hospitalier Régional Universitaire, 59000 Lille, France
Correspondance
Dr. Lilas Al Zein CHU UCL Namur Service d'Anesthésiologie Avenue Gaston Thérasse, 1 B-5530 Yvoir lilas.alzein@student.uclouvain.be
Références
- Druey KM, Parikh SM. Idiopathic systemic capillary leak syndrome (Clarkson disease). J Allergy Clin Immunol. 2016 ; S0091-6749(16):32460-5.
ouvrir dans Pubmed - Ledochowski S, Freichet M, Prieur C, Friggeri A, Lega JC. An uncommon cause of distributive shock: Lessons from two consecutive cases of idiopathic systemic capillary leak syndrome (Clarkson’s disease). Anaesth Crit Care Pain Med. 2015;34(4):251-3.
ouvrir dans Pubmed - Zipponi M, Eugster R, Birrenbach T. High-dose intravenous immunoglobulins : A promising therapeutic approach for idiopathic systemic capillary leak syndrome. BMJ Case Rep. 2011;1220103599.
ouvrir dans Pubmed - Dowden AM, Rullo OJ, Aziz N, Fasano MB, Chatila T, Ballas ZK.: Idiopathic systemi capillary leak syndrome : Novel therapy for acute attacks. J Allergy Clin Immunol. 2009; 124 (5) : 1111-3.
ouvrir dans Pubmed - Duron L, Delestre F, Amoura Z, Arnaud L. Idiopathic and secondary capillary leak syndromes : a systemic review of the literature. Rev Med Interne. 2015; 36(6):386-94.
ouvrir dans Pubmed - Bracq C, Souyris F, Masse C, Boldron A, Poivre P, Vanrenterghem B, et al. Le syndrome d’hyperperméabilité idiopathique : à propos de deux cas, Réanimation. 2001 ; 10 : 232-35.
- Gousseff M, Amoura Z. Idiopathic capillary leak syndrome. Rev Med Interne. 2009; 30(9):754-68.
ouvrir dans Pubmed - Wan XC, Lai A, Kompala T, Ten R. Mimicker of hereditary angioedema: Idiopathic systemic capillary leak syndrome successfully treated with intravenous immunoglobulin. Ann Allergy Asthma Immunol. 2017; 118(5):631-632.
ouvrir dans Pubmed - Lesterhuis WJ, Rennings AJ, Leenders WP, Nooteboom A, Punt CJ, Sweep FC, et al. Vascular Endothelial Growth Factor in Systemic Capillary Leak Syndrome. Am J Med. 2009; 122 (6): 5-7.
ouvrir dans Pubmed - Hirosaki Y, Hayashidani S, Ouchi S, Ohshima T, Nakano R, Yamamoto H. A fatal case of acute progression of generalized edema and simultaneous flash pulmonary edema in a patient with idiopathic systemic capillary leak syndrome: a case report. J Med Case Rep. 2015; 9:90.
ouvrir dans Pubmed - Gousseff M, Arnaud L, Lambert M, Hot A, Hamidou M, Duhaut P, et al. The systemic capillary leak syndrome : a case series of 28 patients from a European registry. Ann Intern Med. 2011;154 (7):464–71.
ouvrir dans Pubmed - Pineton de Chambrun M, Gousseff M, Mauhin W, Lega J-C, Lambert M, Rivière S, et al. Intravenous Immunoglobulins Improve Survival in Monoclonal Gammopathy-Associated Systemic Capillary-Leak Syndrome, Am J Med. 2017; 130 (10): 1219.e19-1219.e27.
ouvrir dans Pubmed - Xie Z, Chan EC, Long LM, Nelson C, Druey KM. High-dose intravenous immunoglobulin therapy for systemic capillary leak syndrome (Clarkson disease). Am J Med. 2015;128(1):91-95.
ouvrir dans Pubmed - Lambert M, Launay D, Hachulla E, Morell-Dubois S, Soland V, Queyrel V, et al. High-dose intravenous immunoglobulins dramatically reverse systemic capillary leak syndrome. Crit Care Med. 2008;36(7):2184–7.
ouvrir dans Pubmed