Introduction
Les symptômes et plaintes hémorragiques ne sont pas inhabituels tant en médecine générale qu’en consultation spécialisée. En effet les épistaxis, les ecchymoses, les hématomes, les saignements post-traumatiques de la vie quotidienne, les gingivorragies, les ménorragies sont des plaintes fréquentes dans la population générale. Leur fréquence est comprise entre 20 et 40% selon les études (1). Cette variabilité est en partie due à la subjectivité des symptômes. Elle peut être atténuée par l’utilisation de scores hémorragiques (bien que ceux-ci ne soient pas toujours utilisés de manière systématique). L’âge auquel ces troubles suscitent une mise au point dépend de leur sévérité. Les troubles mineurs ou modérés (plus fréquents) sont souvent diagnostiqués tardivement, tandis que les troubles plus sévères (plus rares) sont généralement détectés dans l’enfance (2).
L’hémostase est un processus complexe dont le but est d’interrompre les saignements, qu’ils soient spontanés ou provoqués, via la formation d’un thrombus lorsque la paroi vasculaire est endommagée. Ce processus implique une interaction étroite et régulée entre l’endothélium, le tissu conjonctif sous-endothélial (collagène), les plaquettes sanguines, les Facteurs de coagulation ainsi que les acteurs de la fibrinolyse. L’hémostase comprend trois étapes principales : l’hémostase dite primaire, la coagulation et la fibrinolyse. Un dysfonctionnement d’une de ces étapes peut être à l’origine d’une tendance hémorragique anormale. L’identification d’un trouble de l’hémostase devra évaluer chacun de ces acteurs, en tenant compte que le rôle crucial du tissu conjonctif ne peut être évalué au laboratoire (in vitro) (3).
L’évaluation d’une tendance hémorragique anormale requiert une étroite collaboration entre le clinicien et le laboratoire. En effet le caractère subjectif des symptômes exige une anamnèse rigoureuse. Une bonne connaissance des examens réalisables au sein du laboratoire, ainsi que leurs limites, est indispensable. Les résultats biologiques doivent être interprétés en tenant compte des limites des tests réalisés, des caractéristiques du patient et des éventuels résultats d’autres investigations complémentaires (4).
Parmi les indications d’un bilan de coagulation explorant une tendance hémorragique anormale, citons une symptomatologie hémorragique suspecte dans la vie quotidienne, la survenue de complications hémorragiques inattendues lors d’un geste invasif, le dépistage familial d’un déficit connu, la découverte fortuite d’une anomalie d’un test biologique de coagulation lors d’une prise de sang de routine. Une approche standardisée et rigoureuse est recommandée afin d’évaluer les 3 étapes du processus hémostatique. En pratique, il n’est pas rare que malgré un bilan complet, une anomalie biologique pouvant expliquer les symptômes hémorragiques ne soit pas mise en évidence (5).
Une évaluation approfondie permet d’améliorer la prise en charge des patients en leur offrant un traitement hémostatique approprié. Il est important de ne pas étiqueter de manière inappropriée les patients d’une étiologie qui n’est pas formellement établie et d’éviter une exposition inutile à des thérapies hémostatiques (et leurs complications potentielles). De plus, il est essentiel de proposer une prise en charge appropriée pour les patients présentant une anamnèse suspecte avec un bilan biologique normal et non contributif (6).
Cet article se propose de revoir l’évaluation clinique et biologique d’un patient suspecté de présenter une tendance hémorragique anormale.
Interrogatoire
L’interrogatoire systématique revêt une importance primordiale et reste l’outil le plus prédictif. Il est essentiel de tenter de déterminer la localisation des saignements, qu'ils soient muqueux, cutanés, articulaires ou musculaires. Une étiologie plaquettaire ou vasculaire se manifeste typiquement par des saignements muqueux tels que les épistaxis, les gingivorragies, les saignements gynécologiques, digestifs ou urinaires. Un déficit en Facteur de coagulation se caractérise surtout par la survenue d’hémorragies profondes et le développement d’hématomes musculaires ou d’hémarthroses. Cependant, il convient de noter que ces règles générales restent approximatives, comme le déficit en Facteur XI (FXI) qui se caractérise plutôt par des saignements muqueux (7). La chronologie du saignement peut fournir des indications sur une éventuelle anomalie de la fibrinolyse en cas de saignement retardé.
Les éléments les plus prédictifs d’une tendance hémorragique anormale sont les ménorragies, surtout si celles-ci remontent à la ménarche, les saignements anormaux après une chirurgie, les saignements après une extraction dentaire ou lors de blessures mineures, ainsi que les hémorragies du post-partum (8).
L’âge d’apparition revêt une importance capitale, tout comme le caractère spontané ou provoqué des saignements (liés à l’accouchement ou survenant dans le décours d’une intervention chirurgicale, par exemple). Il est également essentiel de rechercher des critères de sévérité tels qu’une anémie, une carence en fer, le recours à une transfusion de globules rouges ou l’administrations de traitements hémostatiques (acide tranexamique, plasma frais, …).
Il convient de recueillir toutes les informations concernant les comorbidités du patient (cirrhose du foie, cancer...), ainsi que la prise de médicaments (anticoagulants, antiplaquettaires, anti-dépresseurs de type SSRI), y compris la prise de médicaments obtenus sans prescription médicale (curcuma, préparations à base de plantes et autres compléments alimentaires). Enfin, il faut rechercher la présence de symptômes suspects de tendance hémorragique anormale au sein de la famille, de considérer l’origine ethnique, de déterminer si la tendance hémorragique affecte les membres de la famille de sexe masculin (hémophilie) et/ou féminin, et de s’assurer de l’absence de consanguinité familiale (pour les maladies récessives non liées au chromosome X) (9).
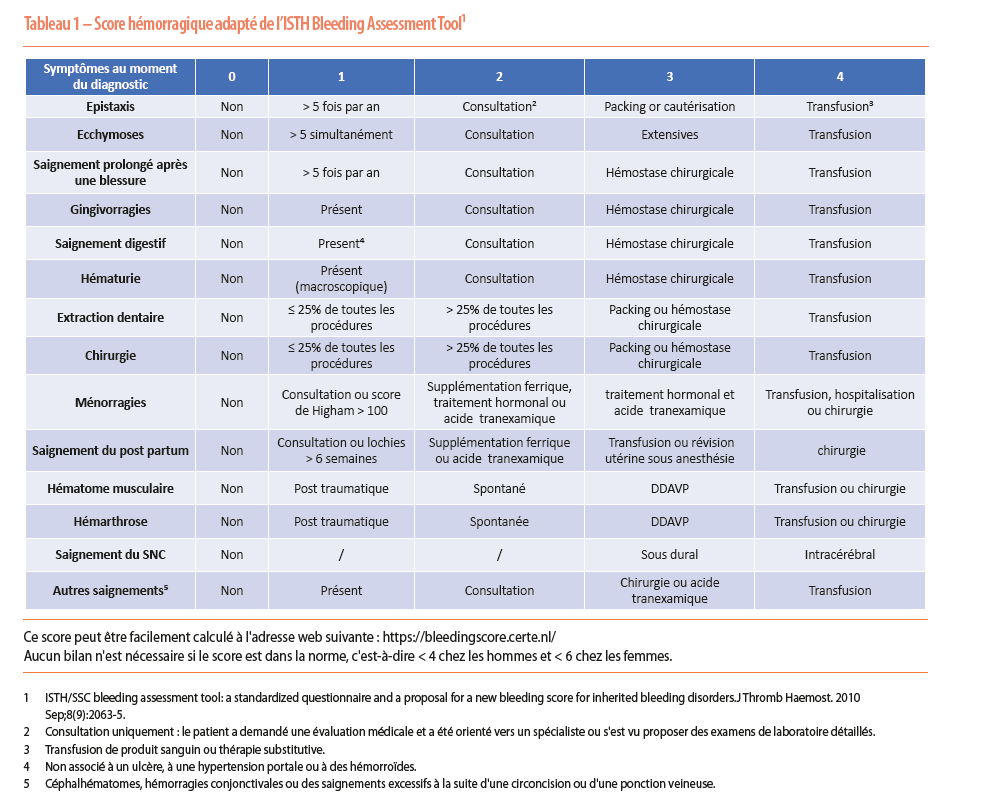
Des scores d’évaluation de la tendance hémorragique doivent idéalement être utilisés pour orienter l’anamnèse, tels que le score ISTH-BAT (Tableau 1). Le résultat doit être interprété avec prudence en tenant compte que ce score a été évalué principalement chez les patients atteints d’une maladie de von Willebrand et qu’il n’est théoriquement pas validé pour les phénotypes modérés et mineurs. Ce score se compose de 14 items évalués en fonction de leur gravité. L’avantage d’un tel score est sa valeur prédictive négative, qui permet d’éviter des examens complémentaires inutiles. En revanche, en cas de score anormal, des investigations supplémentaires seront nécessaires car il ne permet pas de différencier les différentes pathologies. Au plus le score est élevé, au plus la probabilité d’une tendance hémorragique anormale est importante. Ce score ne tient toutefois pas compte des antécédents familiaux hémorragiques. Il est fortement influencé par les « challenges hémostatiques » (opération, accouchements) antérieurs, de telle sorte qu’il est peu sensible chez les jeunes enfants indemnes de tout « challenge » hémostatique (10). L’utilisation préventive de traitements hémostatiques lors de gestes invasifs n’est en outre pas prise en compte. Le score de Higham peut également être utile pour évaluer la sévérité des menstruations.
Paradoxalement, il est également important de s’assurer de l’absence d’antécédents thrombotiques. En effet, certains troubles de la coagulation sont associés à un risque accru de thrombose et de complication obstétricales, tels que les anomalies du fibrinogène, le déficit en Facteur XIII (FXIII), certains déficits en Facteur VII (FVII), les déficits en inhibiteur de l’activateur du plasminogène-1 (PAI-1) ou en alpha-2-antiplasmine (9).
Examen physique
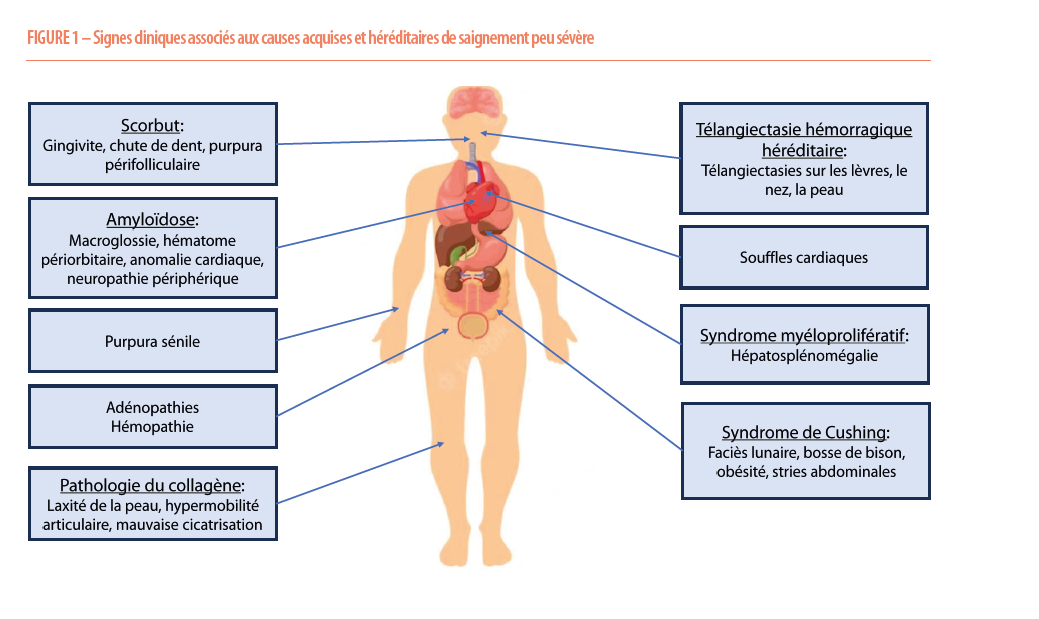
L’examen clinique doit porter une attention particulière à la peau et aux muqueuses, avec la recherche de manifestations hémorragiques comme le purpura, des ecchymoses, des hématomes. La présence de gingivorragie impose un examen des gencives. Il faut évaluer la fragilité cutanée, les signes suspects de mauvaise cicatrisation, ainsi qu’une laxité excessive, suspects de maladie du collagène. Il est important d’être vigilant vis-à-vis de la présence d’angiomes (éventuellement par l’ORL en cas d’épistaxis), qui pourrait faire suspecter une télangiectasie hémorragique héréditaire (maladie d’Osler Rendu, maladie qui affecte une personne sur 5000 soit 2000 patients en Belgique). Les articulations doivent être évaluées à la recherche de déformation, de gonflement ou d’une hyperlaxité. Il faut être attentif vis-à-vis d’autres anomalies morphologiques orientant vers un éventuel syndrome particulier qui pourrait être associé à certains troubles de la coagulation. Enfin en cas d’apparition d’une tendance hémorragique à un âge tardif, il est essentiel d’exclure une cause sous-jacente possible, en recherchant des adénopathies (maladie hématologie maligne sous-jacente), une macroglossie (amyloïdose), des hématomes périorbitaires (suspect d’éventuelle amyloïdose), une hépatosplénomégalie (cirrhose, maladie de surcharge, hémopathie maligne) et un souffle cardiaque (possiblement associé à un déficit acquis en Facteur von Willebrand dégradé par les turbulences anormales occasionnées par une sténose aortique serrée) (6, 7) (Figure 1).
Exploration
Le bilan biologique ne doit être réalisé qu’après une anamnèse rigoureuse, ainsi qu’un examen physique complet. Il comprend une évaluation de l’hémostase primaire, de la coagulation et de la fibrinolyse. Il est important de prendre en compte les limites des différents tests (4). En pratique, nous sollicitons un hémogramme (à la recherche d’une anémie et d’une thrombopénie). Il faut exclure un syndrome inflammatoire (CRP), responsable d’une majoration de certains Facteurs de la coagulation (Facteur von Willebrand, Facteur VIII, raccourcissement du TCA/APTT). Il faut déterminer le groupe sanguin, sachant que les patients du groupe O ont typiquement des concentrations plus basses du Facteur von Willlebrand. Une insuffisance rénale ou hépatique doit être exclue (Tableau 2).
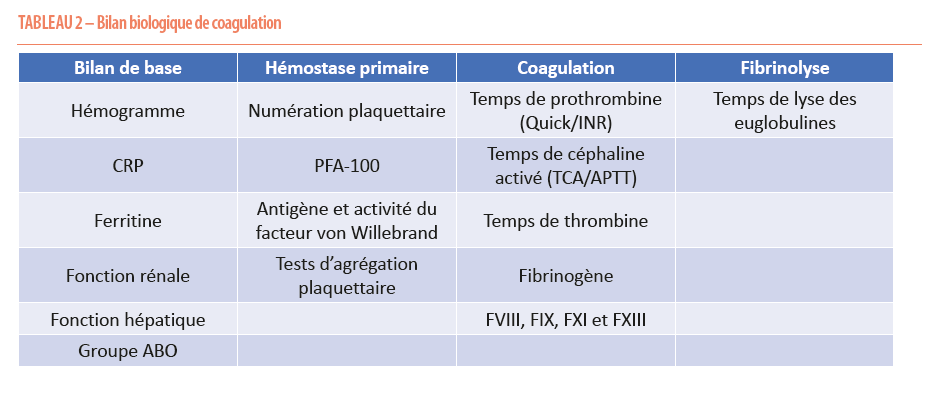
L’hémostase primaire est évaluée par la mesure du PFA (Platelet Function Analyzer-100), un test reproduisant in vitro l’hémostase primaire et sensible aux déficits plaquettaires sévères et en Facteur von Willebrand, les tests d’agrégations plaquettaires (en présence de plusieurs agonistes) et le dosage du Facteur von Willebrand (antigène et activité). La cascade de coagulation est évaluée par les tests de coagulation de base (Temps de prothrombine (TP)/ TCA-APTT/Temps de Thrombine ou TT), en tenant compte de leurs limitations, notamment leur manque de sensibilité pour des déficits peu sévères. La nature des réactifs utilisés a également une influence majeure sur la sensibilité de ces tests aux divers déficits. En d’autres termes, un TCA normal n’exclut pas catégoriquement un déficit partiel, notamment en Facteur VIII (FVIII) et en Facteur IX (FIX). D’autres part les tests de base ne permettent pas de détecter le déficit (exceptionnel) en FXIII (qui intervient dans la stabilisation du caillot en créant des liens chimiques covalents entre les monomères de fibrine). La nature des réactifs utilisés dans les laboratoires et notamment leur richesse en phospholipides influencent la capacité des tests d’hémostase à détecter les déficits en Facteurs et leur sensibilité aux interférences, notamment avec les anticorps anti-phospholipides (anticoagulant du lupus). Ces tests sont par ailleurs insensibles à la fibrinolyse. Pour ces raisons nous demandons systématiquement en plus des tests de base, un dosage du fibrinogène, du FVIII (avec une méthode chromogénique, plus sensible au déficit en FVIII), du FIX, du FXI et du FXIII. Enfin la fibrinolyse est évaluée par la mesure du temps de lyse des euglobulines, même si ce test présente de nombreuses limitations et doit être interprété avec prudence (5).
A l’issue du bilan, un test anormal devra être recontrôlé sur un second prélèvement, afin de ne pas étiqueter erronément un patient d’un déficit qu’il ne présente pas. Par ailleurs, un bilan de base normal (TCA-TP-Fg et TT) n’exclut pas une tendance hémorragique. En cas d’anamnèse douteuse et de score hémorragique anormal, il convient d’explorer l’hémostase primaire, de doser les facteurs de la coagulation et d’évaluer la fibrinolyse. Au terme du bilan, un diagnostic ne sera toutefois établi que parmi 40% des patients. Pour près de 60% des patients présentant une tendance hémorragique anormale, le diagnostic de tendance hémorragique de cause inconnue sera proposé (bleeding of unknown cause, BUC) (11, 12).
Pathologies à suspecter
Troubles acquis
Les troubles acquis de la coagulation sont les plus fréquents. Une bonne anamnèse permettra fréquemment d’établir un diagnostic en évitant un bilan de coagulation inutile.
Iatrogénique
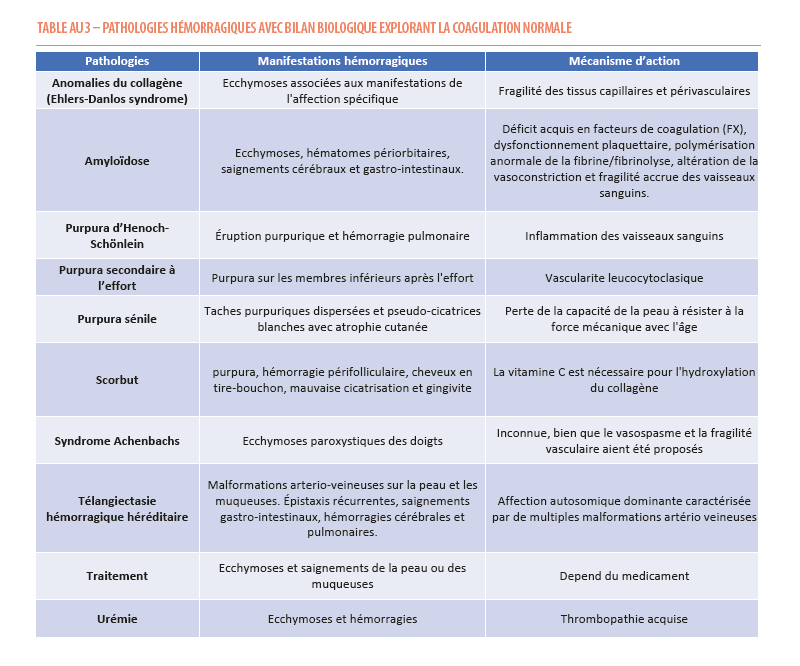
La prise d’un traitement anticoagulant (Sintrom ou autre AVK, anticoagulant oral direct, HBPM…) induit invariablement un risque de saignement plus important. Il en va de même lors de la prescription d’un agent antiagrégant plaquettaire (aspirine, clopidogrel, autres anti-P2Y12). Moins connue des praticiens, la prise d’anti-dépresseurs, tels que les inhibiteurs sélectifs de la recapture de la sérotonine (ISRS) peut induire une thrombopathie mineure, tandis que la prise d’acide valproïque est fréquemment associée à un allongement isolé du temps de thrombine. La prise chronique de corticoïdes, quelle que soit la voie d’administration, peut provoquer une atrophie cutanée et être à l’origine d’hématomes cutanés (9). Enfin, il faut bien interroger le patient à propos de la prise de médicaments non soumis à une prescription médicale, comme le ginkgo biloba, les préparations à base de plantes et autres compléments alimentaires, qui peuvent interférer avec l’hémostase primaire (13). Un bilan d’hémostase n’est par ailleurs pas justifié pour les patients présentant des hématomes liés à une fragilité cutanée secondaire à l’âge (purpura sénile), à la prise de corticoïdes, à la rupture de varicosités des membres inférieurs. L’hématome digital spontané paroxystique (parfois appelé «syndrome d’Achenbach»), une entité méconnue mais fréquente, ne justifie pas de bilan sanguin (Tableau 3). Il se manifeste par une ecchymose soudaine d’un ou de plusieurs doigts, apparaissant spontanément ou après un traumatisme minime et de résolution spontanée (14). Il convient d’interroger les patients à propos de leurs habitudes alimentaires et de s’assurer d’apports suffisants en vitamine C dont le déficit est à l’origine du Scorbut, même si peu fréquent à notre époque mais parfois observé (dénutrition, ..).
Secondaire à une pathologie
L’insuffisance hépatique est fréquemment associée à des anomalies de l’hémostase. En effet, le foie joue un rôle essentiel en produisant à la fois les facteurs de la coagulation, mais également les inhibiteurs de celle-ci tels que les protéine C/S et l’antithrombine. L’insuffisance hépatique n’est classiquement pas associée à un risque hémorragique accru (que pourrait laisser suspecter les anomalies de tests de la coagulation et la thrombopénie) (15). L’insuffisance rénale provoque une thrombopathie qui se traduit par un risque hémorragique accru (16, 17). De manière plus anecdotique le syndrome de Cushing favorise les ecchymoses cutanées, l’amyloïdose peut dans de rare cas majorer le risque hémorragique (par une consommation/absorption du Facteur X et une fibrinolyse accrue). Les pathologies lymphoprolifératives et myéloprolifératives, peuvent être à l’origine d’une hémophilie et d’une maladie de von Willebrand acquises ainsi que d’anomalies plaquettaires (thrombopénie et thrombopathie) (18, 19, 20).
Troubles héréditaires
Tendance hémorragique d’étiologie indéterminée
Malgré une anamnèse suspecte, un diagnostic précis ne sera pas établi chez environ 60% des patients vus en consultation pour tendance hémorragique anormale. Cette prévalence élevée souligne la nécessité de mener des recherches afin d’identifier tous les acteurs impliqués dans le processus d’hémostase (5). D’autre part, certaines anomalies rares responsables d’une tendance hémorragique anormale ont été récemment identifiées mais ne sont pas systématiquement recherchées. Il s’agit du Facteur V court Texas (Atlanta, Amsterdam), des anomalies de la thrombomoduline, des déficits partiels (rares sinon exceptionnels) en Facteur tissulaire (21).
Il est important de ne pas banaliser ces troubles hémorragiques sans étiologie démontrée, car le risque de complications hémorragiques est équivalent à celui d’un patient atteint d’une maladie de von Willebrand prouvée (22). Par conséquent, une prise en charge adaptée doit être proposée afin de prévenir toutes complications lors d’une intervention. L’utilisation d’acide tranexamique (Exacyl®) a fait ses preuves. En cas de saignement incontrôlable, un traitement à base de desmopressine (DDAVP) peut également être envisagé (5, 6).
Les troubles du spectre de l’hypermobilité
Les interactions entre les tissus sous-endothéliaux, le Facteur von Willebrand (FVW) et les plaquettes expliquent que les anomalies du collagène peuvent être associées à une tendance accrue aux saignements. Malheureusement, les tests d’hémostase in vitro de routine ne permettent pas de mesurer l’efficacité de la contribution du collagène sous-endothélial à l’hémostase. Il a été observé que 90% des individus atteints du syndrome d’Ehlers-Danlos (de type hypermobile) présentaient des ecchymoses et des saignements prolongés. En consultation, jusqu’à 20% des patients vu dans le cadre d’un saignement anormal présentent des signes cliniques en faveur d’un syndrome hypermobile ou d’une hyperlaxité (23). Il est donc important de bien interroger les patients à propos des plaintes et symptômes suspects d’anomalies du collagène (entorses, luxations, fragilité cutanée, mauvaise cicatrisation).
La maladie de von Willebrand
La maladie de von Willebrand (VWD) est la maladie hémorragique héréditaire le plus fréquente avec une prévalence estimée à 1% dans la population générale, mais une expression clinique dans 0.01% des cas. Le Facteur de von Willebrand (VWF) est une glycoprotéine plasmatique multimérique indispensable à l’hémostase primaire (adhésion et agrégation des plaquettes) et secondaire (protection du FVIII). La maladie de von VWD est subdivisée en fonction d’un déficit quantitatif partiel (type 1) ou total (type 3), et un déficit qualitatif (type 2). La symptomatologie peut se manifester à tout âge et est très hétérogène. La prise en charge dépendra du contexte et de la sévérité (24). Il est important de rappeler que les personnes du groupe sanguin O présentent des concentrations physiologiquement plus basses du Facteur von Willebrand. Il ne s’agit pas d’une maladie mais d’un déficit lié au groupe O (Low VWF). La concentration du Facteur von Willebrand est influencée par l’âge, le stress, l’inflammation, les œstrogènes (pilule, grossesse) et donc très fluctuante. Il est important de référer les patients vers un centre d’expertise pour confirmation du diagnostic et caractérisation du déficit.
L’hémophilie A et B
Les déficits en FVIII et en FIX sont les déficits en Facteur de coagulation les plus courants dans la population (1000 patients en Belgique). Le phénotype clinique et l’âge de découverte dépendent de la sévérité du déficit. Bien qu’étant une maladie liée à l’X, 30% des patientes sont des femmes qui présentent pour la plupart un déficit peu sévère en FVIII ou FIX (25). La prise en charge a connu d’importantes améliorations ces dernières années, ce qui s’est traduit par une nette augmentation de l’espérance de vie ainsi que de la qualité de vie. Parmi les avancées majeures, mentionnons en particulier le développement d’un agent thérapeutique imitant le FVIII comme l’Hemlibra permettant d’obtenir des taux de FVIII équivalant à 15-20%, grâce à une injection sous cutanée 1 fois par mois. De plus, les traitements à base de FIX à longue durée d’action ont été développés, permettant ainsi une injection intraveineuse tous les 10-14 jours (26). Les patients hémophiles doivent être suivis dans des centres d’expertise. Il n’est pas inhabituel qu’une forme mineure d’hémophilie caractérisée par un déficit en FVIII ou FIX compris entre 5 et 40% soit posé tardivement, de façon fortuite à l’occasion d’un geste invasif ou d’un bilan sanguin.
Déficits en Facteur VII et XI
Il s’agit des déficits en Facteur de coagulation les plus fréquent après l’hémophilie A et B. Ces déficits sont hérités selon un mode de transmission autosomique récessif. Les patients hétérozygotes présentent un phénotype clinique mineur avec des concentrations entre 30 et 70%. La plupart de ces patients doivent recevoir de l’acide tranexamique en cas de geste invasif. Un traitement de substitution ne sera administré qu’en cas de déficit plus sévère, à l’occasion d’un « challenge » hémostatique majeur et en l’absence de réponse à l’acide tranexamique. En cas de déficit sévère en FVII, le traitement recommandé est l’utilisation du Novo Seven® (FVII activé recombinant) à petites doses. En Belgique, l’Hemoleven®, concentré plasmatique de FXI n’est pas disponible en routine, et son administration est associée à un risque thrombotique élevé. Par conséquent, pour corriger un déficit en FXI sévère, du plasma frais congelé sera utilisée (27, 28).
Thrombopathies congénitales
Il s’agit de pathologies rares avec une symptomatologie allant de modérée à sévère. Il peut s’agir d’un déficit d’une des glycoprotéines de surface membranaires, du cytosquelette, d’un déficit granulaire ou enzymatique. Elles peuvent être présentes de manière isolée ou faire partie d’un syndrome, tels que le syndrome de Wiskott-Aldrich ou le syndrome d’Hermansky-Pudlak, ou être associées à une thrombopénie. Les exemples les plus connus sont la thrombasthénie de Glanzmann, le syndrome de Bernard-Soulier et le syndrome des plaquettes grises. En cas d’agrégation plaquettaire anormale, il est recommandé de compléter le bilan par un frottis sanguin, une cytométrie en flux plaquettaire explorant les glycoprotéines de surface, éventuellement associée à une microscopie électronique et un séquençage de nouvelle génération (NGS), en fonction de la sévérité. Les options de traitement sont limitées pour ces pathologies. L’acide transexamique (Exacyl®) et le DDAVP (Minirin®) sont recommandés comme traitements de première ligne. Les transfusions de plaquettes (avec un risque d’allo-immunisation) et l’utilisation de Novo Seven® ne sont préconisées qu’en seconde intention (29).
Prise en charge
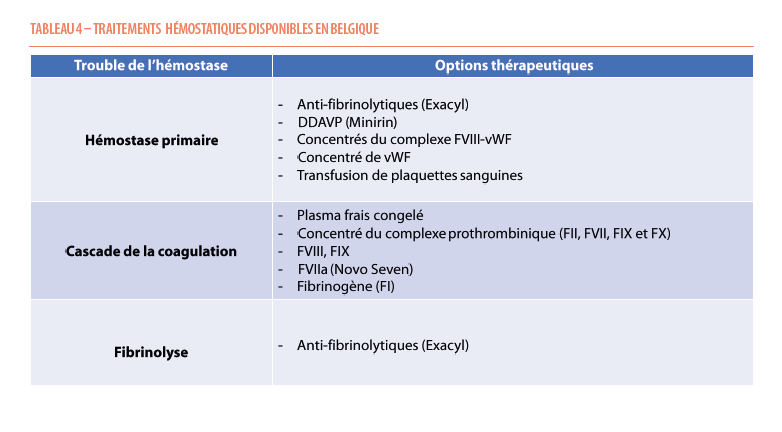
Le traitement hémostatique dépendra, bien entendu de la pathologie sous-jacente ainsi que de sa sévérité. Un traitement sera proposé en fonction des situations, principalement administré ponctuellement à la demande pour prévenir les saignements à l’occasion de gestes invasifs ou administré prophylactiquement (préventivement) de façon régulière afin de limiter les complications à long terme dans les cas les plus sévères (Tableau 4). Il est essentiel que la prise en charge soit multidisciplinaire et spécialisée, impliquant une collaboration entre les différentes spécialités telles que l’ORL, la gynécologie, la chirurgie et l’anesthésie. L’acide tranexamique sera d’une aide précieuse et sera fréquemment utilisé, soit seul en l’absence de déficit objectivé, soit en combinaison avec d’autres approches thérapeutiques en cas de déficit établi et caractérisé (5, 6, 24, 26, 28).
Conclusions
Les troubles de l’hémostase sont variés, pouvant affecter différentes étapes de la coagulation. L’évaluation d’un patient suspect de présenter un tel trouble nécessite une anamnèse détaillée, ainsi qu'un examen physique complet. Le bilan biologique doit évaluer toutes les étapes du processus d’hémostase, tout en tenant compte des éventuelles limitations des tests réalisés. Enfin, il est important de souligner que malgré un bilan bien conduit, un diagnostic n’est établi que dans 40% des cas, les 60% restants étant des troubles de la coagulation d’origine indéterminée qui nécessitent également une prise en charge adaptée.
Affiliations
Service d’hématologie, unité de thrombose et hémostase, Cliniques universitaires Saint-Luc, Université catholique de Louvain (UCLouvain), B-1200 Bruxelles, Belgique
Correspondance
Dr. Michael Iarossi
Université catholique de Louvain
Cliniques universitaires Saint-Luc
Hématologie
Unité de thrombose et hémostase
Avenue Hippocrate 10
B-1200 Bruxelles
Références
- Quiroga T, Mezzano D. Is my patient a bleeder? A diagnostic framework for mild bleeding disorders. Hematology Am Soc Hematol Educ Program. 2012;2012:466-474. doi:10.1182/asheducation.v2012.1.466.3798741
- Mezzano D, Quiroga T. Diagnostic challenges of inherited mild bleeding disorders: a bait for poorly explored clinical and basic research. J Thromb Haemost. 2019;17(2):257-270. doi:10.1111/jth.14363
- Horellou MH, Flaujac C, Gouin Thibault I. Hémostase : physiologie et principaux test d’exploration. EMC - Trait médecine AKOS. 2012;7(2):1-4. doi:10.1016/s1634-6939(12)49382-6
- Hayward CPM. How I investigate for bleeding disorders. Int J Lab Hematol. 2018;40(February):6-14. doi:10.1111/ijlh.12822
- Thomas W, Downes K, Desborough MJR. Bleeding of unknown cause and unclassified bleeding disorders; diagnosis, pathophysiology and management. Haemophilia. 2020;26(6):946-957. doi:10.1111/hae.14174
- Baker RI, O’Donnell JS. How I treat bleeding disorder of unknown cause. Blood. 2021;138(19):1795-1804. doi:10.1182/blood.2020010038
- Boender J, Kruip MJHA, Leebeek FWG. A diagnostic approach to mild bleeding disorders. J Thromb Haemost. 2016;14(8):1507-1516. doi:10.1111/jth.13368
- Hayward CPM. Diagnosis and management of mild bleeding disorders. Hematology Am Soc Hematol Educ Program. 2005;(905):423-428. doi:10.1182/asheducation-2005.1.423
- Rydz N, James P. Approach to the diagnosis and management of common bleeding disorders. Semin Thromb Hemost. 2012;38(7):711-719. doi:10.1055/s-0032-1326783
- Rodeghiero F, Pabinger I, Ragni M, et al. Fundamentals for a Systematic Approach to Mild and Moderate Inherited Bleeding Disorders: An EHA Consensus Report. HemaSphere. 2019;3(5). doi:10.1097/HS9.0000000000000286
- Quiroga T, Goycoolea M, Panes O, et al. High prevalence of bleeders of unknown cause among patients with inherited mucocutaneous bleeding. A prospective study of 280 patients and 299 controls. Haematologica. 2007;92(3):357-365. doi:10.3324/haematol.10816
- Zegers SAM, Smit Y, Saes JL, et al. Diagnostic work up of patients with increased bleeding tendency. Haemophilia. 2020;26(2):269-277. doi:10.1111/hae.13922
- Stanger MJ, Thompson LA, Young AJ, Lieberman HR. Anticoagulant activity of select dietary supplements. Nutr Rev. 2012;70(2):107-117. doi:10.1111/j.1753-4887.2011.00444.x
- Godoy A, Tabares AH. Achenbach syndrome (paroxysmal finger hematoma). Vasc Med (United Kingdom). 2019;24(4):361-366. doi:10.1177/1358863X19849627
- Kujovich JL. Coagulopathy in liver disease: A balancing act. Hematol (United States). 2015;2015(1):243-249. doi:10.1182/asheducation-2015.1.243
- Lambert MP. Platelets in liver and renal disease. Hematology. 2016;2016(1):251-255. doi:10.1182/asheducation-2016.1.251
- van Bladel ER, de Jager RL, Walter D, Cornelissen L, Gaillard CA, Boven LA, et al. Platelets of patients with chronic kidney disease demonstrate deficient platelet reactivity in vitro. BMC Nephrol. 2012;13(1).
- Franchini M, Mannucci PM. Acquired von Willebrand syndrome: Focused for hematologists. Haematologica. 2020;105(8):2032-2037. doi:10.3324/haematol.2020.255117
- Nicol C, Pan-Petesch B, Ianotto JC. Acquired von Willebrand syndrome and lymphoid neoplasms: A review of malignancy management, and propositions of practical recommendations. Haemophilia. 2022;28(6):938-949. doi:10.1111/hae.14648
- Nicol C, Lacut K, Pan-Petesch B, Lippert E, Ianotto JC. Hemorrhage in Essential Thrombocythemia or Polycythemia Vera: Epidemiology, Location, Risk Factors, and Lessons Learned from the Literature. Thromb Haemost. 2021;121(5):553-564. doi:10.1055/s-0040-1720979
- Langdown J, Luddington RJ, Huntington JA, Baglin TP. A hereditary bleeding disorder resulting from a premature stop codon in thrombomodulin (p.Cys537Stop). Blood. 2014;124(12):1951-1956. doi:10.1182/blood-2014-02-557538
- Relke N, Kuthiala S, Grabell J, Hopman WM, James P. The bleeding score: Useful in predicting spontaneous bleeding events in adults with bleeding of unknown cause? Haemophilia. 2020 March ; 26(2): e31–e33. doi:10.1111/hae.13775.
- Jackson SC, Odiaman L, Card RT, Van der Bom JG, Poon MC. Suspected collagen disorders in the bleeding disorder clinic: A case-control study. Haemophilia. 2013;19(2):246-250. doi:10.1111/hae.12020
- Leebeek FWG, Eikenboom JCJ. Von Willebrand’s Disease. N Engl J Med. 2016;375:2067-80. doi:10.1056/NEJMra1601561
- Iorio A, Stonebraker JS, Chambost H, et al. Establishing the prevalence and prevalence at birth of hemophilia in males a meta-analytic approach using national registries. Ann Intern Med. 2019;171(8):542-546. doi:10.7326/M19-1208
- Nogami K, Shima M. Current and future therapies for haemophilia—Beyond factor replacement therapies. Br J Haematol. 2023;200(1):23-34. doi:10.1111/bjh.18379
- Napolitano M, Siragusa S, Mariani G. Factor VII deficiency: Clinical phenotype, genotype and therapy. J Clin Med. 2017;6(4):11-14. doi:10.3390/jcm6040038
- Menegatti M, Peyvandi F. Treatment of rare factor deficiencies other than hemophilia. Blood. 2019;133(5):415-424. doi:10.1182/blood-2018-06-820738
- Kim B. Diagnostic workup of inherited platelet disorders. Blood Res. 2022;57(S1):11-19. doi:10.5045/br.2022.2021223