Introduction
La mucoviscidose est la maladie héréditaire grave la plus fréquente parmi les populations d’origine caucasienne. Transmise sur le mode autosomique récessif, elle implique la présence d’une mutation du gène CFTR (Cystic Fibrosis Transmembrane Regulator) sur chacun des 2 chromosomes 7 de chaque patient. Le gène a été découvert en 1989 et il code pour la protéine du même nom. La maladie affecte en Belgique environ 35 nouveaux nés par an (1/3.000). Dans les pays riches, un traitement symptomatique basé sur une meilleure compréhension de la maladie, a permis depuis les années 1960 une amélioration régulière, de 6 ans par décennie, de l’espérance médiane de vie Au Royaume-Uni, une modélisation de ces progrès continus avait permis de prévoir qu’un nourrisson né en 2.000, diagnostiqué tôt et pris en charge immédiatement dans un bon Centre spécialisé (principal facteur de pronostic) avait une espérance de vie de l’ordre de 50 ans. Cette prédiction a depuis lors été validée (1). La maladie est cependant tellement hétérogène, y compris pour des patients présentant un même génotype CFTR, que ce chiffre déjà dépassé n’a le plus souvent pas de sens pour un patient donné. Le traitement symptomatique actuel est vital, complexe et très lourd : il dévore presque deux heures par jour en moyenne, au très long cours aujourd’hui, ce qui n’a pas d’équivalent à cette échelle dans la médecine. Il est onéreux : en 2012, le coût moyen de la maladie par patient était estimé aux alentours de 45.000 €/an dans des pays comme la France, l’Allemagne ou le Royaume-Uni (2,3). Son efficacité est certaine mais limitée : chez des enfants australiens dépistés en période néonatale et pris en charge immédiatement dans des centres spécialisés, la présence de bronchectasies est quand même documentée dans plus de la moitié des cas dès l’âge de 5 ans (4). L’atteinte pulmonaire conditionne presque toujours le pronostic et en découvrir un traitement plus « fondamental » est donc une nécessité. Un tel traitement ambitionne de contrecarrer le plus tôt possible les conséquences cellulaires de l’anomalie génétique, bien en amont de ce que tente de faire le traitement symptomatique. Idéalement, il s’agit d’un traitement « léger » et bien toléré, à proposer à vie, dès le diagnostic, à des patients aux poumons indemnes qui échapperaient alors aux contraintes de l’actuel traitement symptomatique. La recherche en ce sens est très intense. Les voies envisagées sont multiples (5,6) et se diversifient encore. Parmi les stratégies capables de concerner tous les patients, indépendamment de leur génotype CFTR, se profilent notamment de récentes et fascinantes techniques d’édition de gène (7). Cependant, c’est une approche pharmacologique ciblée en fonction du type de mutations de chaque patient qui est actuellement la plus fructueuse. Il s’agit de développer de petites molécules capables de restaurer au niveau des cellules épithéliales de la paroi des voies aériennes une fonction de la protéine CFTR suffisante pour empêcher les anomalies de microenvironnement ionique menant à un mucus respiratoire anormal, qui va stagner, s’infecter et donner lieu à une inflammation dommageable. De telles molécules sont regroupées sous le terme de « modulateurs ». Dans la population générale, 3 à 4% des personnes sont porteuses d’une mutation du gène CFTR, dont les parents de chaque patient. Ce statut est décrit comme celui de « porteur sain » : la fonction de la protéine CFTR chez eux avoisine 50 % de la fonction normale, sans conséquence respiratoire habituelle. Il est suggéré que restaurer chez les patients une activité de protéine CFTR à hauteur de 25-30 % de la valeur normale suffirait à éviter la cascade d’évènements respiratoires délétères (8). En 2010, la documentation de l’efficacité spectaculaire de l’ivacaftor ( Kalydeco®) chez les rares patients porteurs de la mutation G551D semblait valider cette approche (9). Il apparaît aujourd‘hui réalistement probable que d’ici moins de 5 ans, des associations aussi efficaces de molécules de ce genre (« modulateurs ») pourront être proposées à plus de 90 % des patients (10). Cette perspective constitue l’objet du présent article.
Mutations du gène CFTR : 6 principales classes
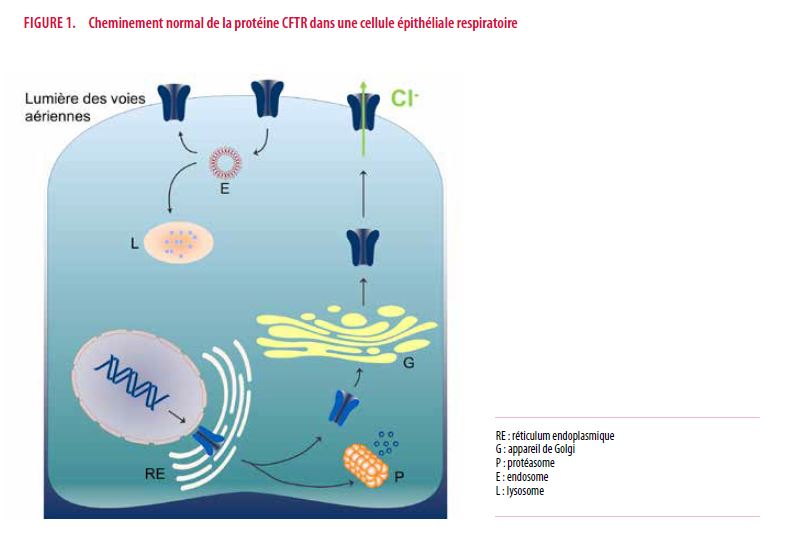
La figure 1 schématise le trajet de la protéine CFTR dans une cellule épithéliale respiratoire normale. Après sa synthèse, la protéine CFTR n’atteint le pôle apical de la cellule, où elle joue notamment le rôle d’un canal pour l’ion chlorure, qu’à la suite d’un processus complexe de maturation intracellulaire impliquant le réticulum endoplasmique et l’appareil de Golgi. Ce processus est soumis à un contrôle de qualité très strict : près de la moitié de la protéine synthétisée est normalement précocement dégradée, principalement par le système protéolytique ubiquitine – protéasome. En outre, le temps de résidence de la protéine fonctionnelle insérée dans la membrane apicale est court. À partir d’endosomes, cette protéine sera réinternalisée pour être soit réinsérée à la membrane, soit dégradée dans des lysosomes. À ce jour, quelque 2.000 mutations putatives du gène CFTR ont été répertoriées. Sur base notamment de données cliniques de plus de 89.000 patients, le projet CFTR2 vise à préciser la portée des plus fréquentes d’entre elles (11). Actuellement, les 400 mutations putatives les plus fréquentes ont été étudiées dans ce cadre (127 sont présentes sur moins de 10 allèles et seulement 78 sur au moins 50 allèles). Vingt mutations putatives (5%) ont été réfutées et reclassées comme des variants sans implications. En Belgique comme globalement au niveau mondial, la seule mutation très fréquente est la mutation F508del : 88% des patients sont chez nous porteurs d’au moins une copie de cette mutation et 48% sont homozygotes (12). Selon leurs conséquences sur la synthèse ou la qualité de la protéine CFTR, les mutations de ce gène sont catégorisées en six classes. Les classes I (absence de synthèse) et II (destruction prématurée et excessive d’une protéine bancale par les mécanismes intracellulaires de contrôle de qualité) ne permettent pas ou pratiquement pas sa présence au pôle apical des cellules. Dans la classe I, certaines mutations (classe Ia) ne permettent pas même de synthèse d’ARN messager. Elles sont parfois reprises sous les termes de Classe VII. Dans la classe III (trouble de régulation), la protéine anormale atteint sa localisation membranaire mais n’est pas fonctionnelle. Dans la classe IV (trouble de conductance), elle l’est partiellement. Dans la classe V (défaut quantitatif), la protéine est fonctionnelle mais sa quantité est trop faible. La classe VI inclut quelques rarissimes mutations donnant lieu à une protéine fonctionnelle mais avec un temps de résidence dans la membrane réduit.
Le tableau I récapitule ces caractéristiques et apporte pour chaque classe quelques exemples de mutations et un ordre de grandeur de la proportion de patients belges concernés (12,13). Les patients porteurs de deux mutations des classes I, II ou III présentent presque toujours une insuffisance pancréatique exocrine, parfois associée à un risque de cirrhose ou de diabète spécifique. Leur atteinte pulmonaire est « globalement » plus évolutive et leurs voies aériennes sont plus souvent chroniquement colonisées par Pseudomonas aeruginosa (14). Les termes de « formes sévères » sont parfois utilisés à leur sujet, par opposition aux « formes légères » que présenteraient les patients porteurs ayant au moins une mutation de classe IV ou V mais cette distinction a peu de sens à l’échelon individuel : le génotype CFTR prédit mal l’évolution respiratoire qui est également tributaire de facteurs d’environnement (dont la qualité des soins) et d’autres facteurs génétiques (gènes modificateurs). Conceptuellement pratique, cette catégorisation en six classes de mutations a plusieurs limites : i) elle est simpliste : une mutation donnée peut entraîner des conséquences qui relèvent de plusieurs classes (cf infra : exemple de la mutation F508del) ; ii) une même conséquence (dégradation accélérée pendant le processus de maturation) peut relever de mécanismes variés et deux mutations de cette classe ne vont pas toujours répondre de la même manière à une même médication (exemple des mutations de classe II F508del et N1303K) iii) la plupart des mutations putatives sont très rares ou rarissimes et leurs conséquences exactes ne sont pas connues de manière précise.
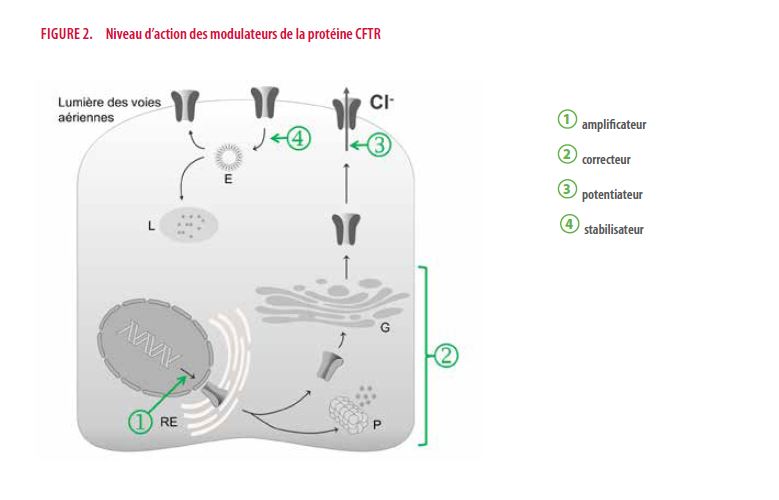
Les modulateurs
Quatre types de modulateurs sont actuellement envisagés, isolément ou en associations.
1. Les amplificateurs visent à augmenter la synthèse brute de protéine CFTR. Dans une toute récente étude randomisée, l’adjonction de PTI-428 chez des adultes homozygotes pour la mutation F508del sous Orkambi® améliore le VEMS de 5% en 4 semaines (15). Dans le cas de mutation de classe Ib, il faut concevoir des molécules capables de permettre la continuité de la synthèse de la protéine malgré la présence d’un signal d’arrêt prématuré (codon stop). L’Ataluren® ne s’en est finalement pas révélé capable (16). D’autres molécules retiennent l’attention (17). L’ELX-02 est en cours d’étude chez la souris (18). Les mutations de classe Ia sont considérées comme hors de portée des modulateurs.
2. Les correcteurs aident à limiter la dégradation excessive d’une protéine bancale avant même qu’elle atteigne la membrane cellulaire. Comme pour le groupe suivant, plusieurs firmes ont à l’étude des molécules de ce genre. Les correcteurs de la firme Vertex sont les plus avancés (lumacaftor, tezacaftor, VX-455, VX-659).
3. Les potentiateurs sont capables d’améliorer la fonction d’une protéine CFTR ayant atteint sa position transmembranaire au pôle apical de la cellule épithéliale. L’ivacaftor (Kalydeco®) de la firme Vertex a été la molécule pionnière de ce type (9) et reste à ce jour la seule commercialisée. Son activité est spectaculaire mais on la sait perfectible. Chez les patients porteurs de la mutation G551D, l’ivacaftor entraîne en quelques semaines une augmentation moyenne du VEMS d’au moins 10% en valeur absolue et une réduction remarquable du taux de chlorure dans la sueur (- 50 mmol/L) qui se normalise dans la moitié des cas. A plus long terme sont documentés d’autres bénéfices comme un ralentissement de moitié de la vitesse de déclin du VEMS, la réduction de plus de 50% de la fréquence des exacerbations pulmonaires, une prise de poids (19,20). L’impact sur la progression des bronchectasies reste à préciser (21,22). Une étude clinique a étendu la portée des résultats cliniques du Kalydeco® aux patients porteurs de huit autres mutations de classe III (23).
4. Un stabilisateur aurait la capacité d’enrayer l’internalisation accélérée de la protéine CFTR transmembranaire et/ou d’en faciliter le recyclage.
La figure 2 illustre le niveau d’intervention de ces modulateurs. La recherche de ces molécules fait appel à des techniques de criblage à haut débit qui ont déjà permis de tester l’intérêt potentiel de plusieurs centaines de milliers de substances. En phase préclinique, diverses méthodes de laboratoire sont utilisées pour confirmer ou non l’intérêt des molécules repérées (études électro-physiologiques sur lignées épithéliales …). D’autres sont en cours de développement, dont les organoïdes (mini-structures digestives ou respiratoires développées à partir de biopsies chez les patients). A noter que si la fonction principale de la protéine CFTR diffère dans l’épithélium respiratoire (efflux de chlorure) et dans l’épithélium de la portion tubulaire excrétrice des glandes sudoripares (réabsorption de chlorure), il existe globalement toutefois dans les études cliniques de modulateurs une certaine corrélation entre l’amélioration de la fonction respiratoire et la diminution du taux de chlorure dans la sueur. Cependant, cette relation est trop lâche pour être utile à une échelle individuelle : chez un patient donné, la variation du taux de chlorure dans la sueur ne permet pas de prédire la réponse en termes de VEMS (24,25).
« Indications » des modulateurs actuels aux USA et en Europe en décembre 2018
Aux USA et en Europe, trois médications de la firme américaine Vertex ont reçu l’agrément des instances de régulation (tableau 2). Les autorisations sont d’abord accordées aux USA et y sont élargies plus tôt à des tranches d’âge plus jeunes. Surtout, les indications agréées y sont plus étendues et l’agréation est rapidement suivie de la mise sur le marché alors qu’en Europe elle ouvre simplement la porte à des négociations entre la firme et chaque pays.
En Europe, le Kalydeco® est remboursé dans plusieurs pays depuis plusieurs années mais l’Orkambi® peine à se frayer un chemin en raison d’un rapport coût/bénéfice médiocre ; le Symkevi® vient à peine d’être agréé. Le rapport coût/bénéfice est davantage pris en considération dans nos pays où la solidarité du système de soins est considérée comme une valeur (cf infra). Les indications potentielles des modulateurs sont plus restrictives en Europe. Agréer une médication très onéreuse sur base seulement d’études précliniques est d’ailleurs questionnable, de même que généraliser une indication pour des mutations classifiées dans le projet CFTR2 comme « à conséquences cliniques variables » telles D1152H et surtout R117H. L’exemple de la mutation R117H, considérée comme principalement une mutation de classe IV à pénétrance variable, est particulièrement parlant. Sa portée est modulée par un polymorphisme de l’intron 8 sur le même gène (variant poly-thymidine). Ce variant peut avoir la forme 5T, 7T ou 9 T. L’association sur le même allèle du variant 5T avec la mutation R117H résulte en un déficit plus marqué de fonction CFTR. Elle est fréquente dans les pays anglo-saxons mais rare en Belgique et en France. Des études françaises ont montré i) que R117H y était la seconde mutation du gène CFTR la plus fréquente dans la population générale ii) qu’elle n’y est associée au variant 5T que dans moins de 5 % des cas iii) que la très grande majorité des personnes porteuses du génotype F508del/R117H n’étaient pas diagnostiquées sans dépistage néonatal et que le risque qu’elles développent à 18 ans une atteinte pulmonaire importante était fort faible (<1%) (26). En conséquence de quoi, cette mutation a depuis lors été retirée du panel de celles recherchées dans le cadre du dépistage néonatal. Par ailleurs, les résultats de l’étude randomisée de l’ivacaftor chez 69 patients porteurs de cette mutation n’ont rien d’euphorique: une amélioration significative du VEMS de l’ordre de 5 % en valeur absolue n’est démontrée que pour le sous-groupe des adultes (n=49), plus malades (27). Le design des études de phase II et III des modulateurs de la firme Vertex est très élaboré et complexe, avec souvent de multiples sous-groupes exposés à des doses différentes. Pour un génotype donné, ces études ne concernent pas nécessairement tous les âges ni tous les stades de sévérité de la maladie. Si l’on s’en tient aux études randomisées de phase III des trois médications agréées, soit le Kalydeco® (9, 23, 28, 29), l’Orkambi® (30-32) et le Symdeko® (33,34), il faut constater la quasi-absence de données chez les patients de plus de 12 ans avec un VEMS supérieur à 90% de la valeur prédite (% pr) ainsi, à tout âge, que l’absence de données concernant les patients les plus malades (VEMS < 40 % pr). Orkambi® et Symdeko® ciblent un marché beaucoup plus large que le Kalydeco®. Chez les patients homozygotes pour la mutation F508del, l’Orkambi® n’entraîne globalement, et à un coût exorbitant (cf infra), qu’une très modeste amélioration du VEMS, de l’ordre de 3%, comparable à celle démontrée dans les années 90 avec un fluidifiant comme la dornase alpha (Pulmozyme®) (35). Ceci n’exclut pas que certains patients en bénéficient plus clairement que d’autres. Une réduction de l’ordre d’un tiers de la fréquence des exacerbations est par ailleurs documentée, qui constitue un bénéfice réel en termes de qualité de vie mais aussi d’évolution de l’atteinte pulmonaire parce qu’un lien est bien établi entre la fréquence des exacerbations et la vitesse de déclin du VEMS (36,37) et qu’on estime qu’environ 20% des exacerbations respiratoires entraînent dans cette maladie une diminution non réversible du VEMS (38,39). Les biomarqueurs traduisent ce modeste bénéfice, avec en particulier une diminution de l’ordre de 10-20 mmol/L du taux de chlorure dans la sueur et une amélioration du transport de chlorure lors de la mesure du potentiel nasal statistiquement significative mais très discrète (- 1. 3 mV) (40). La tolérance de l’Orkambi® est moins bonne lorsque l’atteinte respiratoire est sévère (VEMS < 40% pr) (41-43). Les résultats de l’utilisation d’une médication dans la vie réelle sont toujours moins favorables que ceux des études très encadrées et, en ce qui concerne l’Orkambi®, la plus large étude de ce genre publiée à ce jour (116 patients suivis pendant 11 mois à l’hôpital John’s Hopkins de Baltimore) ne rapporte pas d’amélioration du VEMS sous ce traitement (43). Discrètement plus efficace mais surtout mieux toléré, le Symdeko® rend déjà l’Orkambi® obsolète chez ces patients. De toutes ces limitations, résulte actuellement une certaine confusion qu’illustre bien un essai de recommandations basées sur le niveau de preuves (système GRADE) concernant les indications actuelles des modulateurs par un panel nord-américain de soignants et de patients sans conflit d’intérêt avec l’industrie ni la fondation nord-américaine de la mucoviscidose (44). En dehors du Kalydeco® chez les patients porteurs d’une mutation de classe III, les propositions de ce comité restent limitées, embarrassées et sont souvent explicitement pondérées par la nécessité de prendre en compte le niveau de solvabilité du patient. Enfin, il faut relever que chez les patients porteurs à la fois d’une copie de la mutation F508del et de la mutation G551D (prototype des mutations de classe III), le Symdeko® est plus efficace encore que le Kalydeco®, entraînant une augmentation moyenne du VEMS de 15% plutôt que de 10% (45,46).

Modulateurs de nouvelle génération
Des études in vitro suggèrent que des potentiateurs plus puissants que l’ivacaftor seront découverts (47) et l’idée se précise clairement que ce seront des combinaisons de modulateurs qui apporteront à la plupart des génotypes CFTR une première solution efficace (48). Vu sa fréquence, remédier partiellement au défaut consécutif à la présence de la mutation F508del est un enjeu majeur. Cette mutation est le prototype des mutations de classe II mais si on remédie à la dégradation prématurée de la protéine anormale qui en résulte, cette protéine n’est pas fonctionnelle au niveau de la membrane apicale (classe III) et son temps de résidence y est raccourci (classe VI). L’arrivée de correcteurs de nouvelle génération change la donne. Le tournant paraît bien être le résultat de deux études de phase II publiées cet automne (49,50). Deux de ces nouveaux correcteurs (VX-445 et VX-659) y sont associés en trithérapie à l’ivacaftor et au tezacaftor. Les résultats cliniques apparaissent spectaculaires non seulement chez les patients homozygotes pour la mutation F508del mais également chez les patients porteurs d’une part d’une copie de cette mutation F508del, d’autre part d’une mutation à fonction minimale (Classes I et II). Ce dernier point signifie qu’à terme tous les patients porteurs d’au moins une copie de la mutation F508del seront concernés et on peut anticiper que le bénéfice de ces trithérapies sera supérieur si la seconde mutation appartient aux classe IV et V, et plus encore s’il s’agit d’une mutation de classe III.
La figure 3 résume le bénéfice fonctionnel respiratoire moyen obtenu dans des études randomisées de phase III avec les premiers modulateurs et celui démontré dans les deux études de phase II de trithérapies intégrant des correcteurs de nouvelle génération. Dans ces dernières, à l’issue de quatre semaines de traitement, le VEMS augmente en moyenne de 10 à 14% suivant les groupes. Et, comme avec le Kalydeco® chez les patients porteurs d’une mutation de classe III, cette amélioration fonctionnelle est globalement couplée à une importante réduction du taux de chlorure dans la sueur, de l’ordre de 40 mmol/L. Dans ces études, ces trithérapies n’ont été administrées qu’à un peu plus de 150 patients, tous adultes, et pour une durée limitée. Préalables à leur commercialisation si l’efficacité et la tolérance de ces associations se voient confirmées, six études de phase III concernant ces trithérapies sont déjà en cours. Les quatre études qui concernent des patients d’au moins 12 ans devraient être clôturées en 2019, les deux études concernant des enfants de 6 à 11 ans en janvier 2020 (51).

Plusieurs limitations restent néanmoins à souligner/envisager :
- Si elle est confirmée, une efficacité de cet ordre peut être évaluée simplement et à un échelon individuel comme c’est déjà le cas pour le Kalydeco® mais pour des raisons de pharmacogénétique notamment, certains patients répondront moins que d’autres. On peut à ce propos s’étonner que, s’agissant de médications aussi coûteuses, un monitoring thérapeutique ne soit pas disponible alors que les dosages sont possibles (52).
- En un premier temps, et jusqu’à ce qu’une « désescalade » - qui devrait concerner prioritairement la kinésithérapie respiratoire et les nébulisations - ait pu être étudiée, un tel traitement ne viendra que s’ajouter au traitement symptomatique.
- Ce qui est détruit est irrémédiable : les bronchectasies sévères ne sont pas réversibles et nécessiteront la poursuite d’un traitement symptomatique. Dans quelle mesure leur évolutivité distinctement menaçante dans la mucoviscidose peut être ralentie par ces traitements reste à déterminer. L’importance de la régularité du traitement symptomatique dans les quelques années à venir s’en trouve resoulignée. Même si l’expérience actuelle avec le Kalydeco® suggère que la prévalence de certaines complications de la maladie puisse décroître, elles ne disparaîtront pas, ni les lésions irréversibles d’autres organes que les poumons, comme une cirrhose constituée.
- Pratiquement rien n’est connu des grossesses sous modulateurs, dès lors actuellement contre-indiquées (53).
- Quelques génotypes ne sont pas aidés par la trithérapie décrite. Ainsi en va-t-il notamment des patients porteurs de deux mutations de classe I et probablement aussi de rares patients présentant à l’état homozygote certaines mutations de classe II.
- Etendre les indications aux plus jeunes enfants nécessite des études supplémentaires et donc un peu de temps. Aux USA, la FDA a agréé l’Orkambi® chez l’adulte et l’enfant dès l’âge de 12 ans en juillet 2015, dès 6 ans en septembre 2016, dès 2 ans en août 2018.
- Le coût de ces traitements s’annonce problématique.
À propos du prix des modulateurs
Le nombre des médicaments orphelins augmente rapidement. Ils ont en commun un prix opaque et fort élevé. Globalement, une absence de lien entre d’une part le prix exigé par des firmes souvent en position de monopole et d’autre part les coûts de production, la complexité de la molécule ou le bénéfice thérapeutique a été rapportée (54). Plusieurs auteurs ont souligné cette dérive, comme la nécessité d’une transparence des prix et de la mise en place de mécanismes de régulation (54 – 58). Le coût du Kalydeco® a fait scandale d’emblée, y compris aux USA (59 – 65). L’indice QALY est l’une des mesures utilisées dans l’analyse coût-efficacité d’une médication. Dans les pays médicalisés d’Europe occidentale, le coût annuel qu’une société est habituellement prête à consentir pour un gain d’une année de vie de bonne qualité est souvent estimé entre 20 et 40.000 € (25 à 50.000 $) (66, 67). Il a été calculé que le Kalydeco® pouvait crédiblement ajouter aux patients porteurs d’une mutation de classe III quinze années de vie de bonne qualité, à un coût cependant très élevé, supérieur à 200.000 $ par année (68). Pour l’Orkambi® chez les patients homozygotes pour la mutation F508del, un gain de seulement 2,4 années de vie de bonne qualité est entrevu, au prix de plus d’1.000.000 $/année (69,70). Les médications orphelines vendues par la firme Vertex comptent parmi les plus chères. Tout se passe comme si la firme testait les limites d’un marché très hétérogène : aux USA, les assurances privées acceptent actuellement les prix exigés par Vertex et certains patients qui n’ont pas accès à ces assurances bénéficient d’une aide de la Fondation nord-américaine de mucoviscidose, impliquée dans une saga financière avec la firme (71), mais de tels coûts constituent une menace pour les systèmes de santé solidaires des pays riches d’Europe de l’Ouest et sont tout à fait inaccessibles aux pays plus pauvres (Europe de l’Est, Maghreb, Amérique du Sud …). Le prix des trithérapies entrevues reste à découvrir. Pour les infléchir, la mise en place de nouveaux mécanismes de régulation paraît bien nécessaire (58) et plus sûre que la concurrence qui se met lentement en place sur ce créneau ou l’extension du marché à la grande majorité des patients atteints de mucoviscidose.
Mise en perspective pour le Centre de Référence des Cliniques Saint-Luc
Le Registre Belge de la mucoviscidose montre qu’en 2016, un patient sur 8 - soit 154 patients, pour la plupart (59%) homozygotes pour la mutation F508del (72) - a bénéficié d’une transplantation pulmonaire (12). Les modulateurs n’ont pas d’indication chez ces patients. Au cours des deux dernières années, 210 patients non transplantés ont été vus en consultation à St Luc. Une mutation putative du gène CFTR a pu être identifiée sur 419 de leurs 420 allèles. Si on définit arbitrairement un traitement modulateur spectaculairement efficace comme un traitement capable d’augmenter en moyenne le VEMS d’au moins 10 %, 196 patients (93.4%) dont 187 porteurs d’au moins une copie de la mutation F508del pourraient théoriquement bénéficier d’un tel traitement d’ici cinq ans ou moins, grâce à la trithérapie décrite plus haut (Figure 4). Aujourd’hui, deux tiers de ces 210 patients sont des adultes et deux tiers de ces adultes ont un VEMS supérieur à 70% de la valeur prédite. Quant aux enfants de 6 à 17 ans suivis à Saint-Luc, il y a maintenant 12 ans qu’à la dernière consultation de l’année leur VEMS moyen excède la valeur prédite selon Wang & Hankinson. La grande majorité de cette cohorte ne devrait donc jamais être candidate à une transplantation pulmonaire. Sans compter que de nouveaux modulateurs seront découverts, que l’adjonction d’un amplificateur est une perspective particulièrement prometteuse et que d’autres approches progresseront, dont le bénéfice ne dépend pas du génotype CFTR.

Recommandations pratiques
Des traitements plus fondamentaux et spectaculairement efficaces de l’atteinte pulmonaire qui conditionne presque toujours le pronostic de la mucoviscidose seront bientôt disponibles pour la grande majorité des patients. D’ici-là, l’importance de la régularité du traitement symptomatique actuel s’en trouve encore soulignée.
Affiliations
1 Université catholique de Louvain CHU UCL Namur-Site Godinne Département de Radiologie Avenue Dr G Thérasse 1, 5530, Yvoir, Belgium
2 Institut de recherche expérimentale et clinique, Toxicologie et pharmacologie appliquée. Cliniques universitaires Saint-Luc, Université catholique de Louvain, 10 Avenue Hippocrate 1200 Bruxelles, Belgique
3 Unité de Pneumologie pédiatrique & Mucoviscidose, Cliniques universitaires Saint-Luc, Université catholique de Louvain, 10 Avenue Hippocrate 1200 Bruxelles, Belgique
Correspondance
Pr. Patrick Lebecque, MD, PhD
Université catholique de Louvain
Cliniques universitaires Saint-Luc
Unité de pneumologie pédiatrique et Mucoviscidose
Avenue Hippocrate 10, B-1200 Brussels, Belgium,
Tel.: +32 2 7641939,
Fax: +32 2 7648906
E-mail address: Patrick.Lebecque@uclouvain.be
Références
- Dodge JA, Lewis PA, Stanton M, Wilsher J. Cystic fibrosis mortality and survival in the UK: 1947-2003. Eur Respir J. 2007; 29(3): 522-526.
- Angelis A, Kanavos P, López-Bastida J, Linertová R, Nicod E, Serrano-Aguilar. Research Network. Social and economic costs and health-related quality of life in non-institutionalised patients with cystic fibrosis in the United Kingdom. BMC Health Serv Res. 2015; 15: 428.
- Chevreul K, Michel M, Brigham KB, López-Bastida J, Linertová R, Oliva-Moreno J et al. Social/economic costs and health-related quality of life in patients with cystic fibrosis in Europe. Eur J Health Econ. 2016; 17 Suppl 1: 7-18.
- Stick SM, Brennan S, Murray C, Douglas T, von Ungern-Sternberg BS, Garratt LW et al. Bronchiectasis in infants and preschool children diagnosed with cystic fibrosis after newborn screening. J Pediatr. 2009 ; 155(5): 623-628.
- Cystic Fibrosis Foundation. (https://www.cff.org/Trials/Pipelinepipe-line) (accessed 12/15/2018).
- Cabrini G. Innovative Therapies for Cystic Fibrosis: The Road from Treatment to Cure. Mol Diagn Ther. 2018; doi: 10.1007/s40291-018-0372-6.
- Harrison PT, Hoppe N, Martin U. Gene editing & stem cells. J Cyst Fibros. 2018; 17(1):10-16.
- Zhang L, Button B, Gabriel SE, Burkett S, Yan Y, Skiadopoulos MH et al. CFTR delivery to 25% of surface epithelial cells restores normal rates of mucus transport to human cystic fibrosis airway epithelium. PLoS Biol. 2009; 7(7):e1000155.
- Accurso FJ, Rowe SM, Clancy JP, Boyle MP, Dunitz JM, Durie PR et al. Effect of VX-770 in persons with cystic fibrosis and the G551D-CFTR mutation. N Engl J Med. 2010; 363(21): 1991-2003.
- Clancy JP. Rapid therapeutic advances in CFTR modulator science. Pediatr Pulmonol. 2018; 53(S3): S4-S11.
- CFTR2. https://www.cftr2.org/ (accessed 12/15/2018).
- Annual Data Report Belgian Cystic Fibrosis Registry (BCFR) 2016, Brussels, Belgium
- De Boeck K, Zolin A, Cuppens H, Olesen HV, Viviani L. The relative frequency of CFTR mutation classes in European patients with cystic fibrosis. J Cyst Fibros. 2014; 13(4):403-409.
- McKone EF, Goss CH, Aitken ML. CFTR genotype as a predictor of prognosis in cystic fibrosis. Chest. 2006 ; 130(5): 1441-1447.
- Sawicki G, Pressler T, Schwarz C, Gilmartin G. Phase 2 initial results evaluating PTI-428, a novel CFTR amplifier, in patients with cystic fibrosis. J Cyst Fibros. 2018; 17(S3):S1-2 (abstract).
- Kerem E, Konstan MW, De Boeck K, Accurso FJ, Sermet-Gaudelus I, Wilschanski M et al. Ataluren for the treatment of nonsense-mutation cystic fibrosis: a randomised, double-blind, placebo-controlled phase 3 trial. Lancet Respir Med. 2014; 2(7): 539-547.
- Mutyam V, Du M, Xue X, Keeling KM, White EL, Bostwick JR et al. Discovery of Clinically Approved Agents That Promote Suppression of Cystic Fibrosis Transmembrane Conductance Regulator Nonsense Mutations. Am J Respir Crit Care Med. 2016; 194(9): 1092-1103.
- Rowe SM, Mutyam V, Alroy I, Huertas P. Translational read-through of CFTR nonsense mutations and inducement of cystic fibrosis transmembrane conductance regulator (CFTR) function by ELX-02 treatment. J Cystic Fibros. 2018; 17(S3): s2 (abstract).
- Sawicki GS, McKone EF, Pasta DJ, Millar SJ, Wagener JS, Johnson CA et al. Sustained Benefit from ivacaftor demonstrated by combining clinical trial and cystic fibrosis patient registry data. Am J Respir Crit Care Med. 2015; 192(7): 836-842.
- Bessonova L, Volkova N, Higgins M, Bengtsson L, Tian S, Simard C et al. , Data from the US and UK cystic fibrosis registries support disease modification by CFTR modulation with ivacaftor. Thorax. 2018; 73(8): 731-740.
- Sheikh SI, Long FR, McCoy KS, Johnson T, Ryan-Wenger NA, Hayes D Jr. Computed tomography correlates with improvement with ivacaftor in cystic fibrosis patients with G551D mutation. J Cyst Fibros. 2015; 14(1): 84-89.
- Chassagnon G, Hubert D, Fajac I, Burgel PR, Revel MP; investigators. Long-term computed tomographic changes in cystic fibrosis patients treated with ivacaftor. Eur Respir J. 2016; 48(1):249-52.
- De Boeck K, Munck A, Walker S, Faro A, Hiatt P, Gilmartin G et al. Efficacy and safety of ivacaftor in patients with cystic fibrosis and a non-G551D gating mutation. J Cyst Fibros. 2014 ; 13(6): 674-680.
- Durmowicz AG, Witzmann KA, Rosebraugh CJ, Chowdhury BA. Change in sweat chloride as a clinical end point in cystic fibrosis clinical trials: the ivacaftor experience. Chest. 2013; 143(1): 14-18.
- Fidler MC, Beusmans J, Panorchan P, Van Goor F. Correlation of sweat chloride and percent predicted FEV(1) in cystic fibrosis patients treated with ivacaftor. J Cyst Fibros. 2017 ; 16(1): 41-44.
- Thauvin-Robinet C, Munck A, Huet F, Génin E, Bellis G, Gautier E et al. The very low penetrance of cystic fibrosis for the R117H mutation: a reappraisal for genetic counselling and newborn screening. J Med Genet. 2009; 46(11): 752-758.
- Moss RB, Flume PA, Elborn JS, Cooke J, Rowe SM, McColley SA et al. Efficacy and safety of ivacaftor in patients with cystic fibrosis who have an Arg117His-CFTR mutation: a double-blind, randomised controlled trial. Lancet Respir Med. 2015; 3(7): 524-533.
- Ramsey BW, Davies J, McElvaney NG, Tullis E, Bell SC, Dřevínek P et al. A CFTR potentiator in patients with cystic fibrosis and the G551D mutation. N Engl J Med. 2011; 365(18): 1663-1672.
- Davies J, Sheridan H, Bell N, Cunningham S, Davis SD, Elborn JS et al. Assessment of clinical response to ivacaftor with lung clearance index in cystic fibrosis patients with a G551D-CFTR mutation and preserved spirometry: a randomised controlled trial. Lancet Respir Med. 2013; 1(8): 630-638.
- Boyle MP, Bell SC, Konstan MW, McColley SA, Rowe SM, Rietschel E et al. A CFTR corrector (lumacaftor) and a CFTR potentiator (ivacaftor) for treatment of patients with cystic fibrosis who have a phe508del CFTR mutation: a phase 2 randomised controlled trial. Lancet Respir Med. 2014; 2(7): 527-538.
- Wainwright CE, Elborn JS, Ramsey BW, Marigowda G, Huang X, Cipolli M et al. Lumacaftor-Ivacaftor in Patients with Cystic Fibrosis Homozygous for Phe508del CFTR. N Engl J Med. 2015; 373(3): 220-231.
- Ratjen F, Hug C, Marigowda G, Tian S, Huang X, Stanojevic S et al. Efficacy and safety of lumacaftor and ivacaftor in patients aged 6-11 years with cystic fibrosis homozygous for F508del-CFTR: a randomised, placebo-controlled phase 3 trial. Lancet Respir Med. 2017; 5(7): 557-567.
- Taylor-Cousar JL, Munck A, McKone EF, van der Ent CK, Moeller A, Simard C et al. Tezacaftor-Ivacaftor in Patients with Cystic Fibrosis Homozygous for Phe508del. N Engl J Med. 2017; 377(21): 2013-2023.
- Rowe SM, Daines C, Ringshausen FC, Kerem E, Wilson J, Tullis E et al. Tezacaftor-Ivacaftor in Residual-Function Heterozygotes with Cystic Fibrosis. N Engl J Med. 2017; 377(21): 2024-2035.
- Fuchs HJ, Borowitz DS, Christiansen DH, Morris EM, Nash ML, Ramsey BW et al. Effect of aerosolized recombinant human DNase on exacerbations of respiratory symptoms and on pulmonary function in patients with cystic fibrosis. The Pulmozyme Study Group. N Engl J Med. 1994; 331(10): 637-642.
- de Boer K, Vandemheen KL, Tullis E, Doucette S, Fergusson D, Freitag A et al. Exacerbation frequency and clinical outcomes in adult patients with cystic fibrosis. Thorax. 2011; 66(8):680-685.
- Sanders DB, Bittner RC, Rosenfeld M, Redding GJ, Goss CH. Pulmonary exacerbations are associated with subsequent FEV1 decline in both adults and children with cystic fibrosis. Pediatr Pulmonol. 2011; 46(4):393-400.
- Sanders DB, Bittner RC, Rosenfeld M, Hoffman LR, Redding GJ, Goss CH. Failure to recover to baseline pulmonary function after cystic fibrosis pulmonary exacerbation. Am J Respir Crit Care Med. 2010; 182(5):627-632.
- Heltshe SL, Goss CH, Thompson V, Sagel SD, Sanders DB, Marshall BC et al. Short-term and long-term response to pulmonary exacerbation treatment in cystic fibrosis. Thorax. 2016; 71(3):223-229.
- Graeber SY, Dopfer C, Naehrlich L, Gyulumyan L, Scheuermann H, Hirtz S et al. Effects of Lumacaftor-Ivacaftor Therapy on Cystic Fibrosis Transmembrane Conductance Regulator Function in Phe508del Homozygous Patients with Cystic Fibrosis. Am J Respir Crit Care Med. 2018; 197(11):1433-1442.
- Popowicz N, Wood J, Tai A, Morey S, Mulrennan S. Immediate effects of lumacaftor/ivacaftor administration on lung function in patients with severe cystic fibrosis lung disease. J Cyst Fibros. 2017; 16(3):392-394.
- Hubert D, Chiron R, Camara B, Grenet D, Prévotat A, Bassinet L et al. Real-life initiation of lumacaftor/ivacaftor combination in adults with cystic fibrosis homozygous for the Phe508del CFTR mutation and severe lung disease. J Cyst Fibros. 2017; 16(3):388-391.
- Jennings MT, Dezube R, Paranjape S, West NE, Hong G, Braun A et al. An Observational Study of Outcomes and Tolerances in Patients with Cystic Fibrosis Initiated on Lumacaftor/Ivacaftor. Ann Am Thorac Soc. 2017; 14(11):1662-1666.
- Ren CL, Morgan RL, Oermann C, Resnick HE, Brady C, Campbell A et al. Cystic Fibrosis Foundation Pulmonary Guidelines. Use of Cystic Fibrosis Transmembrane Conductance Regulator Modulator Therapy in Patients with Cystic Fibrosis. Ann Am Thorac Soc. 2018; 15(3):271-280.
- Donaldson SH, Pilewski JM, Griese M, Cooke J, Viswanathan L, Tullis E et al. Tezacaftor/Ivacaftor in Subjects with Cystic Fibrosis and F508del/F508del-CFTR or F508del/G551D-CFTR. Am J Respir Crit Care Med. 2018; 197(2):214-224.
- Guimbellot J, Sharma J, Rowe SM. Toward inclusive therapy with CFTR modulators: Progress and challenges. Pediatr Pulmonol. 2017; 52(S48):S4-S14.
- Gees M, Musch S, Van der Plas S, Wesse AS, Vandevelde A, Verdonck K et al. Identification and Characterization of Novel CFTR Potentiators. Front Pharmacol. 2018; 9: 1221.
- Carlile GW, Yang Q, Matthes E, Liao J, Radinovic S, Miyamoto C et al. A novel triple combination of pharmacological chaperones improves F508del-CFTR correction. Sci Rep. 2018; 8(1):11404.
- Keating D, Marigowda G, Burr L, Daines C, Mall MA, McKone EF et al. VX-445-Tezacaftor-Ivacaftor in Patients with Cystic Fibrosis and One or Two Phe508del Alleles. N Engl J Med. 2018; 379(17): 1612-1620.
- Davies JC, Moskowitz SM, Brown C, Horsley A, Mall MA, McKone EF et al. VX-659-Tezacaftor-Ivacaftor in Patients with Cystic Fibrosis and One or Two Phe508del Alleles. N Engl J Med. 2018; 379(17): 1599-1611.
- https://clinicaltrials.gov/ct2/search (accessed 12/15/2018)
- Schneider EK, Reyes-Ortega F, Wilson J, Kotsimbos T, Keating D, Velkov T, Li J. Development of a LC-MS/MS method for the analysis of ivacaftor, its major metabolites and lumacaftor in plasma and sputum of cystic fibrosis patients treated with Orkambi or Kalydeco. J Cyst Fibros. 2017; 16 S1: S24 (abstract).
- Heltshe SL, Godfrey EM, Josephy T, Aitken ML, Taylor-Cousar JL. Pregnancy among cystic fibrosis women in the era of CFTR modulators. J Cyst Fibros. 2017; 16(6): 687-694.
- Roos JC, Hyry HI, Cox TM. Orphan drug pricing may warrant a competition law investigation. BMJ. 2010; 341: c6471.
- Simoens S. Pricing and reimbursement of orphan drugs: the need for more transparency. Orphanet J Rare Dis. 2011; 6: 42.
- Hughes-Wilson W, Palma A, Schuurman A, Simoens S. Paying for the Orphan Drug System: break or bend? Is it time for a new evaluation system for payers in Europe to take account of new rare disease treatments? Orphanet J Rare Dis. 2012; 7: 74.
- Picavet E, Morel T, Cassiman D, Simoens S. Shining a light in the black box of orphan drug pricing. Orphanet J Rare Dis. 2014; 9: 62.
- Luzzatto L, Hyry HI, Schieppati A, Costa E, Simoens S, Schaefer F et al. Outrageous prices of orphan drugs: a call for collaboration. Lancet. 2018; 392(10149): 791-794.
- Bush A, Simmonds NJ. Hot off the breath: ‘I’ve a cost for’ - the 64 million dollar question. Thorax. 2012; 67(5): 382-384.
- https://www.medpagetoday.com/upload/2013/5/17/CFletter.pdf (accessed 12/15/2018)
- Barrett PM, Alagely A, Topol EJ. Cystic fibrosis in an era of genomically guided therapy. Hum Mol Genet. 2012; 21(R1): R66-71.
- O’Sullivan BP, Orenstein DM, Milla CE. Pricing for orphan drugs: will the market bear what society cannot? JAMA. 2013; 310(13): 1343-1344.
- Cohen D, Raftery J. Paying twice: questions over high cost of cystic fibrosis drug developed with charitable funding. BMJ. 2014; 348: g1445.
- Balfour-Lynn IM. Personalised medicine in cystic fibrosis is unaffordable. Paediatr Respir Rev. 2014; 15 Suppl 1: 2-5.
- Ferkol T, Quinton P. Precision Medicine: At What Price? Am J Respir Crit Care Med. 2015; 192(6): 658-659.
- Owens DK. Interpretation of cost-effectiveness analyses. J Gen Intern Med. 1998; 13(10): 716-717. Review.
- Simoens S. How to assess the value of medicines? Front Pharmacol. 2010; 1: 115.
- Dilokthornsakul P, Hansen RN, Campbell JD. Forecasting US ivacaftor outcomes and cost in cystic fibrosis patients with the G551D mutation. Eur Respir J. 2016; 47(6): 1697-1705.
- Dilokthornsakul P, Patidar M, Campbell JD. Forecasting the Long-Term Clinical and Economic Outcomes of Lumacaftor/Ivacaftor in Cystic Fibrosis Patients with Homozygous phe508del Mutation. Value Health. 2017; 20(10): 1329-1335.
- Sharma D, Xing S, Hung YT, Caskey RN, Dowell ML, Touchette DR. Cost-effectiveness analysis of lumacaftor and ivacaftor combination for the treatment of patients with cystic fibrosis in the United States. Orphanet J Rare Dis. 2018; 13(1): 172.
- Orenstein DM, Abood RN. Cost(s) of caring for patients with cystic fibrosis. Curr Opin Pediatr. 2018; 30(3): 393-398.
- Thomas M, Wanyama S, Dupont L, Etienne I, Evrard P, Rondelet B et al. Demography and clinical outcomes in cystic fibrosis lung transplant recipients in Belgium. J Cyst Fibros. 2018; 17, S3: s47 (abstract).