POLYKYSTOSE RÉNALE AUTOSOMIQUE DOMINANTE – ENFIN UN TRAITEMENT FREINANT SA PROGRESSION !
Nathalie Demoulin, Anh Ho, Nathalie Godefroid, Caroline Clerckx, Laurence Annet, Yves Pirson, Olivier Devuyst
INTRODUCTION
La polykystose rénale autosomique dominante (PKRAD) est la néphropathie héréditaire la plus fréquente, atteignant une personne sur 400-2000. La PKRAD est la cause de l’insuffisance rénale terminale chez environ 10% des patients dialysés ou transplantés rénaux, soit environ 50.000 personnes en Europe. Causée par une mutation dans un des gènes PKD1 ou PKD2, la maladie se caractérise par le développement de multiples kystes rénaux, conduisant à une augmentation progressive de la taille des reins et à une destruction du parenchyme rénal. Plus de la moitié des patients atteignent le stade d’insuffisance rénale terminale avant l’âge de 65 ans. Les atteintes extra-rénales de la PKRAD sont principalement l’hypertension artérielle, le développement de kystes hépatiques (surtout chez la femme) et les anévrismes intracrâniens.
LES NOUVEAUX TRAITEMENTS DE LA POLYKYSTOSE
Jusqu’il y a peu, la prise en charge de la PKRAD reposait exclusivement sur le traitement néphroprotecteur conventionnel, en privilégiant l’utilisation d’inhibiteurs du système rénine-angiotensine-aldostérone pour traiter l’hypertension artérielle (1). Les avancées dans la connaissance de la physiopathologie de la PKRAD ont permis le développement de traitements ciblés, bloquant les voies de signalisation impliquées dans la progression de la néphropathie. Ainsi, le tolvaptan, un antagoniste sélectif du récepteur V2 de la vasopressine (ou hormone antidiurétique) dans le canal collecteur, diminue la concentration d’AMPcyclique intracellulaire, un médiateur central des phénomènes impliqués dans la formation et la progression des kystes. L’étude randomisée TEMPO 3:4 (à laquelle les Cliniques universitaires Saint-Luc ont participé très activement) a bien montré que le tolvaptan réduisait de moitié la progression de la croissance du volume rénal par rapport au placébo, sur une période d’observation de 3 ans. Le tolvaptan a également permis de freiner la progression de l’insuffisance rénale, avec une diminution de 30% du déclin de la fonction rénale.
Les sous-analyses de l’étude TEMPO ont montré que le bénéfice du tolvaptan était similaire selon le degré d’insuffisance rénale à l’inclusion (débit de filtration glomérulaire ≥90 vs 60-89 vs 30-59 ml/min/1.73m², ce dernier groupe ne représentant que moins de 20% des patients) (2). Par ailleurs, le bénéfice du tolvaptan était supérieur chez les patients présentant une suppression plus importante de l’osmolalité urinaire, ce qui souligne la pertinence du lien entre la voie de signalisation médiée par le récepteur V2 et la progression de la PKRAD (3). Les patients sous tolvaptan présentaient significativement moins de douleurs rénales, d’épisodes d’hématurie macroscopique et d’infections urinaires (4).
Sans surprise, les effets secondaires présentés par les patients sous tolvaptan reflètent son action aquarétique : pollakiurie, nycturie et soif. On a relevé une toxicité hépatique chez 5% des patients sous tolvaptan, réversible à l’arrêt du traitement, et sans facteur de risque identifié.
L’Agence Européenne des Médicaments (AEM) a approuvé en février 2015 l’utilisation du tolvaptan chez les « patients avec PKRAD rapidement progressive ayant une fonction rénale normale ou modérément réduite (débit de filtration glomérulaire ≥30 ml/min/1.73m2) », moyennant un suivi étroit de l’enzymologie hépatique (voir ci-dessous).
Parmi les autres traitements en cours d’évaluation, les analogues de la somatostatine (e.g. lanréotide) paraissent prometteurs. Ils sont déjà utilisés ‘off-label’ pour le traitement de la polykystose hépatique. Les Cliniques universitaires Saint-Luc ont acquis une importante expérience de ce traitement.
DE NOUVEAUX SCORES POUR L’IDENTIFICATION DES PATIENTS À HAUT RISQUE DE DÉCLIN FONCTIONNEL RÉNAL
Par rapport à l’augmentation du volume rénal, l’altération de la fonction rénale est tardive dans l’évolution de la PKRAD. Le déclin du débit de filtration glomérulaire n’a donc une valeur pronostique qu’à un stade avancé de la maladie. Tout récemment, deux nouveaux scores pronostiques ont été développés et validés dans de grandes cohortes :
1. Un score clinico-génétique (PROPKD score) basé sur le sexe, la survenue de l’hypertension ou d’une première complication urologique avant 35 ans, et le type de mutation PKD1/PKD2 en cause. Le diagnostic moléculaire de la PKRAD devient plus accessible grâce aux techniques de séquençage à haut débit. La variabilité intra-familiale parfois assez franche du phénotype rénal oblige cependant à postuler l’existence de facteurs additionnels (génétiques ou environnementaux) qui ne sont pas pris en compte dans ce score.
2. Un score basé sur le volume rénal total (VRT) à un âge donné, mesuré par résonance magnétique ou par scanner, et corrigé pour la taille. Le développement d’une méthode (ellipsoïde) pratique de quantification du VRT a rendu accessible cette technique chez les patients ayant une forme typique de polykystose, avec un temps d’analyse de 7 minutes, au lieu des 40 minutes nécessaires pour la planimétrie manuelle (gold standard). La vitesse de croissance du VRT (en moyenne, 5% par an) peut aussi être utilisée pour prédire l’évolution de la fonction rénale et s’avère donc un outil pronostique performant et accessible.
Ces nouveaux scores pronostiques, complétés par des marqueurs tels que l’osmolalité urinaire (Uosm) (3), nous permettent d’identifier les patients à haut risque de progression rapide, qui seront le plus à même de bénéficier des traitements disponibles. Ils ont d’ailleurs été repris dans les recommandations émises de l’ERA-EDTA (Société Européenne de Néphrologie) concernant l’identification des patients porteurs d’une PKRAD candidats au traitement par tolvaptan (5).
LE TOLVAPTAN, REMBOURSÉ EN BELGIQUE EN 2016
Le Jinarc® (tolvaptan) est remboursé en Belgique depuis début septembre 2016 pour le traitement des patients porteurs d’une polykystose rénale autosomique dominante, âgés de 18-50 ans, ayant un VRT > 750ml, un débit de filtration glomérulaire ≥ 30 ml/min/1.73m2 et à risque de progression rapide d’après le VRT corrigé pour la taille et à un âge donné (classification de la Mayo Clinic 2015: http://www.mayo.edu/research/documents/pkd-center-adpkd-classification/d...). La demande de remboursement (initiale et de renouvellement semestriel) doit être introduite par un néphrologue attaché à un hôpital universitaire. Le suivi mensuel de l’enzymologie hépatique est obligatoire pendant 18 mois puis 1x/3mois.
STANDARDISATION DE LA PRISE EN CHARGE DES PATIENTS
Afin d’optimiser et de standardiser la prise en charge des patients porteurs de PKRAD, un panel d’experts internationaux, dont les professeurs Devuyst et Pirson, et de patients concernés par la PKRAD se sont réunis sous l’égide de KDIGO (Kidney Disease Improving Global Outcomes), qui publie les guidelines mondiales en néphrologie. Leurs recommandations en termes de prise en charge clinique et suggestions de domaines de recherche ont été publiées récemment (1). La création d’équipes multidisciplinaires prenant en charge les complications des patients (polykystose hépatique majeure, anévrismes cérébraux, douleurs chroniques…) est encouragée. Nous nous y employons aux Cliniques Saint-Luc.
QUELQUES MOTS SUR LA PKRAD CHEZ LES ENFANTS
La PKRAD a toujours été considérée comme une maladie de l’adulte alors que les kystes se développent déjà en anténatal, avec une vitesse de croissance qui dépasse largement celle observée chez l’adulte. On a longtemps pensé que les kystes n’entrainaient pas de symptômes chez l’enfant. Il apparaît cependant que les manifestations de la maladie peuvent être précoces. Ainsi, 25 à 30% des enfants avec PKRAD présentent une hypertension artérielle, 23% une protéinurie et 10% une hématurie (6). Ces enfants peuvent développer une hypertrophie ventriculaire gauche même s’ils sont normotendus. Il semblerait qu’ils présentent en premier lieu une hypertension artérielle nocturne avec une perte du dip nocturne. Les enfants développant la maladie en anténatal ou avant 18 mois semblent évoluer vers une forme plus sévère. Il n’existe actuellement aucun consensus sur l’opportunité de tester et de suivre les enfants à risque de développer une PKRAD. Il faudrait pour ce faire pouvoir démontrer qu’un dépistage précoce et systématique dès l’enfance, et la mise en route de mesures thérapeutiques adaptées influencent positivement l’évolution au long cours de la maladie. Une étude multicentrique internationale évaluant la tolérance et l’efficacité du tolvaptan chez l’enfant à partir de 4 ans a débuté en septembre 2016, les Cliniques universitaires Saint-Luc y prendront part.
En résumé, la PKRAD est une maladie menant à l’insuffisance rénale terminale chez bon nombre de patients et ayant un impact majeur sur leur qualité de vie. Le tolvaptan permet de ralentir la croissance kystique et donc le déclin fonctionnel rénal. Il est disponible en Belgique depuis septembre 2016 pour les patients à progression rapide. Nous disposons de plusieurs outils pronostiques permettant d’identifier les patients les plus susceptibles de bénéficier des nouveaux traitements. La nécessité de tester et de suivre les enfants à risque de développer cette maladie n’a pas encore été démontrée mais le suivi de la tension artérielle semble important. Le développement de centres d’expertise et de cliniques multidisciplinaires s’avère indispensable à la prise en charge optimale et individualisée de chaque patient atteint de PKRAD.
RÉFÉRENCES
1. Chapman AB, Devuyst O, Eckardt KU, Gansevoort RT, Harris T, Horie S, et al. Autosomal-dominant polycystic kidney disease (ADPKD): executive summary from a Kidney Disease: Improving Global Outcomes (KDIGO) Controversies Conference. Kidney Int 2015; 88: 17-27.
ouvrir dans Pubmed
2. Torres VE, Higashihara E, Devuyst O, Chapman AB, Gansevoort RT, Grantham JJ, et al. Effect of Tolvaptan in Autosomal Dominant Polycystic Kidney Disease by CKD stage: Results from the TEMPO 3:4 Trial. Clin J Am Soc Nephrol 2016; 11: 803-811.
ouvrir dans Pubmed
3. Devuyst O, Chapman AB, Gansevoort RT, Higashihara E, Perrone RD, Torres VE, et al. Urine Osmolality, Response to Tolvaptan, and Outcome in Autosomal Dominant Polycystic Kidney Disease: results from the TEMPO 3:4 Trial. J Am Soc Nephrol 2016.
ouvrir dans Pubmed
4. Casteleijn NF, Blais JD, Chapman AB, Czerwiec FS, Devuyst O, Higashihara E, et al. Tolvaptan and Kidney Pain in Patients with Autosomal Dominant Polycystic Kidney Disease: Secondary Analysis from a Randomized Controlled Trial. Am J Kidney Dis 2017; 69: 210-219.
ouvrir dans Pubmed
5. Gansevoort RT, Arici M, Benzing T, Birn H, Capasso G, Covic A, et al. Recommendations for the use of tolvaptan in autosomal dominant polycystic kidney disease: a position statement on behalf of the ERA-EDTA Working Groups on Inherited Kidney Disorders and European Renal Best Practice. Nephrol Dial Transplant 2016; 31: 337-348.
ouvrir dans Pubmed
6. Reddy BV, Chapman AB. The spectrum of autosomal dominant polycystic kidney disease in children and adolescents. Pediatr Nephrol 2017; 32: 31-42.
ouvrir dans Pubmed
AVANCÉES DANS LA GLOMÉRULONÉPHRITE EXTRA-MEMBRANEUSE IDIOPATHIQUE : RÔLE DE L’AUTO-IMMUNITÉ ET CIBLES ANTIGÉNIQUES
Michel Jadoul, Selda Aydin, Sylvie Goletti, Johann Morelle, Nathalie Demoulin
Le syndrome néphrotique est défini par une triade : protéinurie abondante (> 3.5 g/24 h, ce qui correspond en pratique clinique à 4+ à la bandelette ou > 3g de protéines/g de créatinine sur un échantillon d’urine), hypoalbuminémie (< 3 g/dL) et œdèmes. Ce syndrome signe l’existence d’une maladie glomérulaire, et peut être causé par le diabète, l’amylose AL (associée ou non au myélome multiple), le lupus érythémateux disséminé, et d’autres maladies affectant spécifiquement les reins. Hormis en cas de néphropathie diabétique évidente, le diagnostic étiologique d’un syndrome néphrotique de l’adulte nécessite la réalisation d’une biopsie rénale et une analyse histologique détaillée.
La glomérulonéphrite extra-membraneuse (GEM), caractérisée en immunofluorescence par des dépôts de complexes immuns sur le versant externe de la membrane basale glomérulaire (Figure), constitue la cause la plus fréquente de syndrome néphrotique idiopathique de l’adulte.
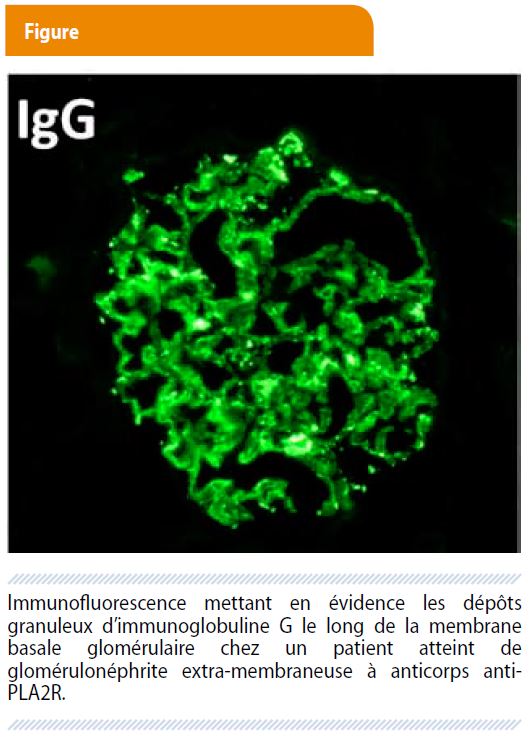
L’origine auto-immune de la GEM idiopathique était suspectée depuis plusieurs décennies sur base de modèles expérimentaux (néphrite de Heymann chez le rat), à côté des formes secondaires associées au virus de l’hépatite B, à certains cancers ou à des médicaments. L’origine auto-immune de la GEM idiopathique fut démontrée par le groupe de David Salant à Boston en 2009, avec la mise en évidence d’une première cible antigénique. Cette équipe a ainsi identifié le récepteur de la phospholipase A2, un antigène exprimé à la surface des podocytes glomérulaires humains, comme cible des auto-anticorps constitutifs des complexes immuns déposés chez environ 70% des patients atteints de GEM idiopathique. Plusieurs travaux récents (1) ont démontré que ces anticorps circulants anti-PLA2R ont une bonne spécificité pour la GEM idiopathique auto-immune. Le titre d’anticorps anti-PLA2R prédit en outre l’évolution de la maladie (peu de rémissions spontanées si le titre d’anti-PLA2R est élevé) et la réponse au traitement immunosuppresseur (association de corticostéroides et cyclophosphamide, cyclosporine, tacrolimus, voire rituximab). De façon intéressante, une diminution voire une négativation du titre d’anti-PLA2R, spontanée ou sous immunosuppresseurs, précède de plusieurs mois l’amélioration de la protéinurie. Enfin, comme dans d’autres affections auto-immunes, le phénomène de spreading moléculaire vient d’être rapporté pour les anticorps anti-PLA2R : un changement d’épitope(s) cible(s) des anti-PLA2R au cours de l’affection va de pair avec une évolution défavorable (2).
Une équipe française a récemment identifié un autre antigène podocytaire, la Thrombospondine D7A (THSD7A), comme cible alternative des auto-anticorps dans les GEM idiopathiques. Les anti-THSD7A sont beaucoup plus rares (3%) que les anticorps anti-PLA2R, et principalement détectés chez des femmes. Cependant, deux points concernant les anti-THSD7A méritent d’être soulignés. Premièrement, dans un cas de GEM avec présence d’anticorps anti-THSD7A, une rémission de la maladie a été obtenue après le traitement par chimiothérapie d’un carcinome de la vésicule biliaire, diagnostiqué simultanément (3). L’examen immunohistochimique a montré une expression abondante de THSD7A au sein de la tumeur biliaire et des métastases ganglionnaires, mais pas dans le tissu sain avoisinant. Il est dès lors possible qu’une partie des GEM idiopathiques à anti-THSD7A soient en réalité paranéoplasiques. Ensuite, une observation clinique toute récente a montré, qu’un titre élevé d’anti-THSD7A au moment de la transplantation rénale était associé à une récidive de GEM sur le greffon rénal. L’antigène THSD7A étant exprimé par les podocytes glomérulaires murins, les auteurs ont en outre pu montrer que l’injection d’anticorps anti-THSD7A humains chez la souris induit des lésions rénales typiques de GEM et une protéinurie. Cette observation conforte le rôle pathogène direct des anticorps anti-THSD7A (4).
En conclusion, après des décennies de stagnation, la recherche consacrée aux GEM idiopathiques progresse très rapidement, avec des retombées cliniques évidentes. Ces développements modifient profondément la prise en charge des patients atteints de GEM idiopathique, du bilan étiologique initial au suivi régulier ultérieur. Aux Cliniques universitaires Saint-Luc, le dosage des anticorps circulants anti-PLA2R et la recherche des dépôts glomérulaires de PLA2R sont effectués en routine, par ELISA (méthode quantitative) et par immunohistochimie, respectivement. La recherche des anticorps circulants anti-THSD7A (par immunofluorescence indirecte, méthode semi-quantitative) (5) et de l’antigène correspondant sur la biopsie devrait suivre sous peu.
RÉFÉRENCES
1. Cattran DC, Brenchley PE. Membranous nephropathy : integrating basic science into improved clinical management. Kidney Int 2017, Jan 5 : pii: S0085-2538(16)30617-2. doi: 10.1016/j.kint.2016.09.048.
ouvrir dans Pubmed
2. Seitz-Polski B, Dolla G, Payré C, Girard CA, Polidori J, Zorzi K et al. Epitope Spreading of Autoantibody Response to PLA2R Associates with Poor Prognosis in Membranous Nephropathy. J Am Soc Nephrol 2016; 27: 1517-1533.
ouvrir dans Pubmed
3. Hoxha E, Wiech T, Stahl PR, Zahner G, Tomas NM, Meyer-Schwesinger C, et al. A Mechanism for Cancer-Associated Membranous Nephropathy. N Engl J Med 2016; 374: 1995-1996.
ouvrir dans Pubmed
4. Tomas NM, Hoxha E, Reinicke AT, Fester L, Helmchen U, Gerth J, et al. Autoantibodies against thrombospondin type 1 domain-containing 7A induce membranous nephropathy. J Clin Invest 2016; 126: 2519-2532.
ouvrir dans Pubmed
5. Hoxha E, Beck LH Jr, Wiech T, Tomas NM, Probst C, Mindorf S, et al. An Indirect Immunofluorescence Method Facilitates Detection of Thrombospondin Type 1 Domain-Containing 7A-Specific Antibodies in Membranous Nephropathy. J Am Soc Nephrol 2017; 28: 520-531.
ouvrir dans Pubmed
LE PROGRAMME D’ÉCHANGE DE REINS DE DONNEURS VIVANTS POUR LA TRANSPLANTATION RÉNALE : L’EXPÉRIENCE EN BELGIQUE
Eric Goffin, Michel Mourad
La transplantation rénale est le traitement de choix de l’insuffisance rénale terminale, permettant une meilleure qualité de vie et une espérance de vie allongée aux patients urémiques, en comparaison à la dialyse. La transplantation rénale se fait à partir d’un rein de donneur décédé ou à partir d’un rein de donneur vivant, apparenté ou non. Malheureusement, il existe depuis de nombreuses années une pénurie de reins de donneurs décédés (1) partout en Europe si bien que la transplantation de rein de donneur vivant est encouragée chaque fois que cela est possible. Les avantages d’une greffe de rein à partir d’un donneur vivant sont nombreux : pour le receveur, possibilité d’une intervention pré-emptive, c’est-à-dire avant le début de la dialyse, certitude de recevoir un rein d’excellente qualité et de bénéficier d’une préparation chirurgicale et immunologique extensive, et instauration éventuelle du traitement immunosuppresseur avant l’intervention chirurgicale ; les bénéfices pour le donneur sont principalement d’éviter ou d’interrompre la dialyse chez le receveur, mais également d’ordre moral (estime de soi, service rendu …). En outre, le nombre de patients en liste d’attente pour une transplantation rénale par rein de donneur décédé va ainsi être réduit. Cependant, dans certains cas, le candidat-donneur ne peut être retenu en raison d’une incompatibilité dans les groupes sanguins ABO ou en raison de la présence d’anticorps anti-HLA chez le receveur dirigés contre son donneur. C’est pour permettre de retenir la candidature de ces donneurs récusés au don de rein qu’un programme d’échanges de reins a été mis sur pied dans la plupart des programmes de greffe actuels dans le monde (2-3). Le principe en est extrêmement simple : le donneur A ne peut donner son rein à son receveur A ; il en va de même dans le couple B. Il est dès lors proposé au donneur A de donner son rein au receveur B alors que le donneur B donne en même temps son rein au receveur A. Des chaînes d’échanges plus complexes peuvent aussi être bâties selon les caractéristiques des couples en présence selon un modèle informatique complexe élaboré à Eurotransplant (fig) (4). Bien sûr, toutes ces propositions d’échanges respectent les critères cliniques et immunologiques élémentaires requis pour une transplantation d’organe.


Les premiers échanges de reins de donneurs vivants dans le monde basés sur ces principes ont été réalisés dès 2004 aux Pays-Bas (5) ; les néerlandais ayant effectués à ce jour 263 transplantations de reins « échangés » entre donneurs vivants. Des programmes actifs similaires existent actuellement aux USA, Canada, Australie, Corée et dans la plupart des pays européens ; ces programmes sont encouragés par les sociétés nationales de transplantation et par les autorités de santé. Une chaine « record » permettant la réalisation de 30 greffes a d’ailleurs été décrite aux USA par le New York Times en 2012 (6).
Et en Belgique ? Un programme similaire d’échange de reins de donneurs vivants est également actif depuis quelques années. Des travaux préliminaires sur le sujet avaient débuté dès Septembre 2006 sous l’égide de la Société Belge de Transplantation avec finalisation d’un protocole complet en Novembre 2007. Ce protocole, accepté par les 7 centres belges de transplantation, est en accord total avec la législation belge sur le don de rein à partir d’un donneur vivant. L’analyse des possibilités d’échanges de rein entre les différents couples, ce qui est appelé un « run », se fait à Eurotranplant. Si un échange entre 2 couples est possible, les donneurs et receveurs sont hospitalisés dans leur centre de greffe respectifs et les reins des donneurs sont acheminés de l’hôpital « préleveur » vers l’hôpital « greffeur » (les distances en Belgique étant relativement courtes, le temps d’ischémie du greffon n’est jamais très long). Evidemment les procédures de prélèvement, et donc de greffe, doivent être effectuées de façon simultanée dans les 2 hôpitaux. Les médecins responsables du programme dans chaque hôpital doivent s’assurer de l’anonymat des donneurs et receveurs, ainsi que du caractère équitable de l’échange afin de garantir un maximum de chances de succès aux 2 paires. Enfin, il n’y a pas de compensation financière prévue pour les donneurs et receveurs impliqués.
Un certain nombre de principes de base ont également été ajoutés dans ce programme pour en assurer la pérennité :
1. favoriser un maximum d’échanges entre couples à chaque « run » ;
2. favoriser des échanges avec groupes sanguins identiques (A vers A, O vers O) plutôt que compatibles (O vers A,..) ;
3. priorité est donnée aux patients avec probabilité faible de trouver une compatibilité HLA ;
4. favoriser les chaines courtes ;
5. ne pas dépasser une différence d’âges > 15 ans entre les couples.
L’évaluation des donneurs et receveurs se fait dans chaque centre transplanteur selon les procédures habituelles de ce centre. Il est possible à chaque centre participant de demander des renseignements cliniques, immunologiques … supplémentaires pour s’assurer de l’équité de l’échange et même, de refuser l’échange proposé, le cas échéant. Les patients, donneur et receveur, doivent signer un consentement éclairé au moment de l’inscription au programme d’échange en présence d’un témoin. Les patients candidat-receveurs sont habituellement inscrit en liste d’attente Eurotransplant pour une greffe à partir d’un rein de donneur décédé en même temps qu’ils participent au programme d’échange.
Les premiers essais d’échanges en Belgique ont eu lieu en Avril 2013 et impliquaient 6 couples ; depuis 2015, 3 à 4 « run » sont effectués annuellement et impliquent habituellement 18 à 20 couples. Lors de la dernière analyse, les raisons de participation au programme d’échange étaient une incompatibilité ABO et la présence d’anticorps spécifiques contre le receveur dans 56 et 44 % des cas, respectivement. Les donneurs étaient le conjoint (77 %), un parent (7 %), un frère ou une sœur (8 %), un beau-frère (4 %) ou un ami (4 %).
À ce jour, 7 greffes ont pu être réalisées grâce à ce système d’échange : 2 fois un échange simple entre 2 couples et une fois, un échange entre 3 couples. Le temps d’ischémie a varié entre 2 heures 31 minutes pour la procédure la plus courte et 4 heures 5 minutes, pour la plus longue. Aucune complication n’est survenue. Cinq greffons sont actuellement parfaitement fonctionnels ; deux greffons ont malheureusement été perdus à la suite de multiples rejets. Ce programme d’échange de reins à partir de donneurs vivants est donc une expérience intéressante pour augmenter les possibilités de greffe rénale en Belgique. Ce challenge n’est possible que grâce à une collaboration excellente entre tous les centres belges de greffe, et avec Eurotransplant. Une information régulière des médecins généralistes et néphrologues, des autorités de la santé et du grand public est indispensable pour faire connaître et développer ce programme.
RÉFÉRENCES
1. https://www.eurotransplant.org/cms/. Eurotransplant annual report 2016.
2. Rapaport FT. The case for a living emotionally related international kidney donor exchange registry. Transplant Proc 1986 ; 18 (Suppl. 2) : 5-9.
ouvrir dans Pubmed
3. Ferrari P, Weimar W, Johnson RJ, Lim WH, Tinckam KJ. Kidney paired donation: principles, protocols and programs. Nephrol Dial Transplant 2015 ; 30 : 1276-1285.
ouvrir dans Pubmed
4. De Klerk M, Van Der Deijl WM, Witvliet MD, Haase-Kromwijk BJ, Claas FH, Weimar W. The optimal chain length for kidney paired exchanges: an analysis of the Dutch program. Transpl Int 2010 ; 23 : 1120-1225.
ouvrir dans Pubmed
5. De Klerk M, Keizer KM, Claas FH, Witvliet M, Haase-Kromwijk BJ, Weimar W. The Dutch national living donor kidney exchange program. Am J Transplant 2005 ; 5 : 2302-5.
ouvrir dans Pubmed
6. Sack K. Sixty lives, 30 kidneys, all linked. Ney York Times Feb 18, 2012 : 2302-2305.
Affiliations
Correspondance
Pr. Michel Jadoul
michel.jadoul@uclouvain.be
Pr. Eric Goffin
eric.goffin@uclouvain.be
Dr. Nathalie Demoulin
nathalie.demoulin@uclouvain.be