Le syndrome VEXAS (Vacuoles, Enzyme E1, X-linked, Autoinflammatory, Somatic) est une pathologie inflammatoire de l’adulte causée par la mutation somatique du gène UBA1 dans les cellules hématopoïétiques. Les patients présentent des symptômes inflammatoires, rhumatologiques et hématologiques. Actuellement, en l’absence de traitement efficace, la morbidité est importante et la mortalité précoce. Combinant une clinique inflammatoire et hématologique, le VEXAS se présente comme le prototype d’une nouvelle classe de maladie « hémato-inflammatoire ».
Historique
Le syndrome VEXAS a été décrit pour le première fois en décembre 2020 par Beck et son équipe (1). Les auteurs ont utilisé une approche inédite basée sur l’analyse génotypique de patients dans le but de mettre en évidence une maladie phénotypique. Dans le cas présent, le génome de plus de 2500 patients présentant des symptômes inflammatoires systémiques sans diagnostic précis. (fièvres récurrentes, vasculites et éruptions cutanées diverses) a été analysé, ce qui a permis de mettre en évidence chez 25 de ces patients une mutation récurrente au niveau du gène UBA1. Les patients concernés étaient des hommes âgés de plus de 40 ans. Cette mutation n’était pas connue des bases de données génomiques. Elle est retrouvée au niveau des cellules hématopoïétiques, exclusivement au niveau des cellules myéloïdes. Aucun cas familial n’a été décrit. La mise en évidence d’une inflammation systémique précoce associée à une mortalité importante dans un modèle expérimental in vivo de zébra-fish permet de confirmer l’hypothèse des auteurs d’un lien de cause à effet entre la mutation UBA1 et la clinique inflammatoire présentée par ces patients.
Physiopathologie
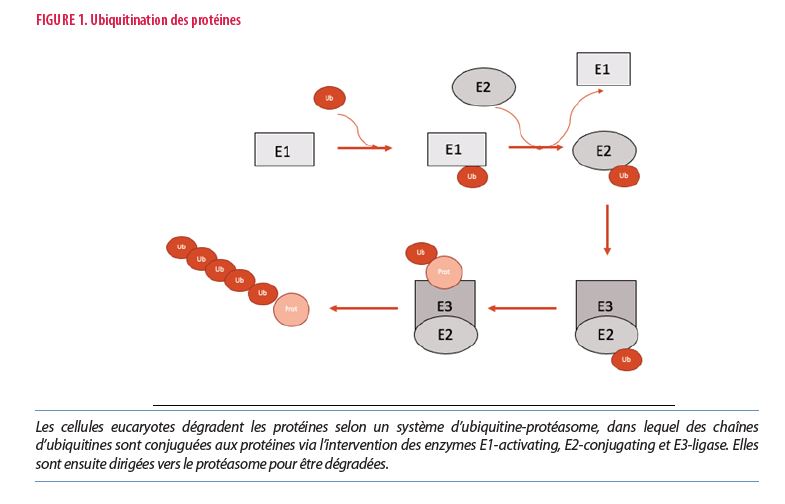
Le gène UBA1 code pour l’enzyme E1, première enzyme à initier le cycle d’ubiquitination des protéines au niveau intracellulaire. Pour rappel, les cellules eucaryotes dégradent les protéines selon un système d’ubiquitine-protéasome, dans lequel des chaînes d’ubiquitine sont conjuguées aux protéines afin qu’elles soient dirigées et ensuite dégradées dans le protéasome (Figure 1). L’ubiquitination est un processus complexe, faisant schématiquement intervenir 3 familles d’enzymes dont l’enzyme E1-activating qui permet l'initiation du cycle en activant l’ubiquitine. Les enzymes E2-conjugating et E3-ligase permettent leur conjugaison et leur liaison aux protéines, respectivement. Contrairement aux enzymes E2 et E3 qui possèdent plusieurs dizaines d’isoformes, il n’existe que 2 isoformes d’UBA1 : une isoforme nucléaire dont la transcription est initiée à la première méthionine (Met), et une isoforme cytoplasmique initié à la 41ème méthionine. Une fois les protéines totalement ubiquitinisées, elles sont dirigées vers le protéasome où elles sont finalement détruites.
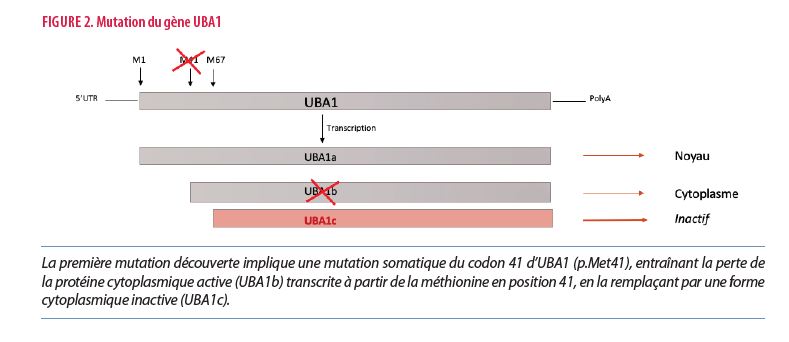
Les premières mutations découvertes, et les plus fréquentes selon les données actuelles, implique une mutation somatique de la méthionine en position 41 du gène UBA1, entraînant la perte de la protéine cytoplasmique active en la remplaçant par une forme cytoplasmique inactive (UBA1c) transcrite à partir de la méthionine en position 67 (Figure 2). Dans ce type de mutation, la méthionine 41 est alors remplacée par une valine (p.Met41Val : c.121A->G), une thréonine (p.Met41Thr : c.122T->C) ou une leucine (p.Met41Leu : c.121A->C). Depuis la description du syndrome VEXAS, d’autres variants plus rares commencent à être décrits. Ces isoformes inactives s’accumulent au niveau du cytoplasme, entraînant un stress cellulaire responsable de l’apoptose de la cellule et de la libération de cytokines inflammatoires dans le milieu extracellulaire. L’hypothèse qui prévaut actuellement est que le microenvironnement des cellules myéloïdes mutées devient inflammatoire et favorise la survenue de mutations pouvant entraîner des maladies clonales médullaires. À noter que le gène UBA1 se trouve sur le chromosome X, expliquant la polarisation masculine de la pathologie. Cependant, des cas féminins de VEXAS commencent à être décrits selon l’inactivation aléatoire d’un de leur chromosome X, avec cependant une clinique généralement moins marquée.
Diagnostic
Le diagnostic est génétique. Il consiste à mettre en évidence la mutation du gène UBA1 par méthode Sanger ou par NGS (next generation sequencing). La méthode Sanger nécessite un mosaïcisme cellulaire d’au moins 15% chez les patients VEXAS pour permettre une détection de la mutation, mais elle reste la méthode de premier choix pour des raisons de rapidité et de coût. En cas de forte suspicion clinique malgré un test de Sanger négatif, un examen par NGS doit être réalisé, sa sensibilité étant de l’ordre du pourcent. Des études ultérieures devront cependant discriminer la relevance clinique des quelques pourcents de mosaïcisme retrouvés chez certains patients.
Manifestations cliniques
Les patients sont pour la plupart des hommes de plus de 40-50 ans, souffrant de manifestations cliniques inflammatoires depuis plusieurs années. On peut retrouver des fièvres récurrentes, des atteintes pulmonaires (pneumopathie interstitielle), articulaires (arthrite, chondrite nasale et auriculaire) ou encore cutanées (dermatose neutrophilique, vasculite) (2-3). Pour rappel, des femmes peuvent présenter des formes atténuées de la pathologie.
Manifestations hématologiques (4)
Étant donné que la mutation du gène UBA1 se retrouve au niveau des cellules myéloïdes, cela entraîne des implications non seulement au niveau de l’hémogramme et du médullogramme, mais également au niveau de la propension à développer des maladies hématologiques malignes.
Au niveau de l’hémogramme et du frottis sanguin, une macrocytose est retrouvée chez tous les patients atteints de VEXAS. Celle-ci peut être associée à la présence de précurseurs granulocytaires immatures circulants, de neutrophiles et monocytes vacuolés, et également de neutrophiles hypogranulaires ou hyposegmentaires.
L’aspect caractéristique est retrouvé au niveau du médullogramme, où l’on observe la présence de vacuoles au niveau des précurseurs hématopoïétiques myéloïdes chez tous les patients. Ces vacuoles ne sont pas spécifiques du VEXAS et peuvent également être retrouvées dans la carence en cuivre, l’excès de zinc, la consommation chronique d’alcool et les pathologies myéloïdes ou lymphoïdes au sens large. À noter qu’il faut également exclure des artéfacts apparus lors de l’étalement de la moelle. Ces vacuoles doivent cependant attirer l’attention du biologiste si elle touche des précurseurs très jeunes tels que des érythroblastes et des promyélocytes, si elle est associée à une hypercellularité et à un contexte clinique suggestif d’une dérégulation au niveau du système inflammatoire.
En raison de la présence de ce microenvironnement inflammatoire au niveau médullaire et d’une possible expansion clonale primaire due à la prolifération et/ou à l’avantage de survie des cellules mutées, il n’est pas surprenant de découvrir au fil des études que ces patients sont plus à risque de développer des pathologies hématologiques clonales. On retrouve ainsi de la myélodysplasie chez 25 à 55% des patients selon les études (1, 5-6), de la dyscrasie plasmocytaire (myélome multiple et MGUS) chez 12,5% (4) et des lymphocytoses monoclonales B chez 12,5% (4).
Un risque thrombo-embolique majoré a également été décrit avec le VEXAS, principalement veineux, mais des cas de thromboses artérielles existent également (7). Ce risque s’explique par une activation de la cascade de la coagulation par les cytokines inflammatoires, la présence d’une inflammation au niveau des vaisseaux sanguins dans le contexte de vasculite, ainsi qu’à un rôle probable de l’anticoagulant du lupus qui peut être retrouvé chez près de 44% des patients atteints de VEXAS. Ces thromboses peuvent survenir sans autre facteur de risque associé, parfois de façon répétée chez certains patients, voire même parfois en présence d’une anticoagulation bien conduite.
Ces chiffres sont à prendre avec précaution en raison du faible nombre de patients rapportés par les études à l’heure actuelle, mais les données ultérieures permettront d’affiner ces pourcentages. Il est cependant essentiel pour tous patients ayant un diagnostic de VEXAS d’avoir un suivi hématologique rapproché.
Pronostic et traitement
Bien que la description génétique du VEXAS permette à ces patients d’avoir un diagnostic précis, leur survie reste faible avec un taux de mortalité entre 25 à 35% à 5 ans (6-8), qui cependant s’affine au fur et à mesure des nouvelles données. Ainsi, une étude impliquant 73 patients présentée au récent congrès américain de rhumatologie a permis de mettre en évidence la présence du variant valine et la transfusion-dépendance des patients comme facteurs prédictifs de mortalité. Les syndromes VEXAS associés à un variant valine ont une mortalité de 50%, beaucoup plus importante que les variants leucine (18%) et thréonine (22%) (8). La médiane de survie d’un VEXAS à variant valine est de 9 ans après le début des symptômes (8).
Dans un avenir proche, le génotype permettra de déterminer les futurs choix de traitement qui restent actuellement limités. La cortico-résistance est fréquente, et les symptômes sont mal contrôlés malgré de nombreuses lignes d’immunosuppresseurs. Plusieurs pistes thérapeutiques sont actuellement étudiées dont les anticorps monoclonaux anti-IL6 et les inhibiteurs de JAK (janus kinase)-2 (dont le Ruxolitinib pour lequel une étude est actuellement en cours) qui semblent les plus prometteurs. L’allogreffe de cellules souches hématopoïétiques est la seule option curative actuellement (9), mais limitée devant l’âge et la fragilité de ces patients pouvant avoir des comorbidités associées. Elle reste réservée aux patients jeunes, sans comorbidité, et probablement recommandée en cas de variant valine et/ou de dépendance aux transfusions.
Conclusions
Le VEXAS est un syndrome associant une clinique inflammatoire et des anomalies hématologiques chez des hommes (et plus rarement des femmes) de plus de 40-50 ans. Le diagnostic est génétique, basé sur la mise en évidence par méthode Sanger ou NGS d’une mutation du gène UBA1 dans les cellules hématopoïétiques. La morbi-mortalité est élevée, avec une cortico-résistance fréquente. Les traitements les plus prometteurs sont les anticorps monoclonaux anti-IL6 et les inhibiteurs des JAK2, actuellement à l’étude. L’allogreffe de cellules souches hématopoïétiques doit être discutée chez les patients jeunes, porteurs d’un variant valine et/ou dépendants des transfusions.
Depuis la découverte de ce syndrome, plus de 350 cas ont été décrits dans la littérature (7-8), souvent de manière rétrospective. De nombreux nouveaux cas de VEXAS sont régulièrement décrits permettant d’améliorer notre compréhension de cette pathologie « hémato-inflammatoire » afin de proposer une prise charge optimale de ces patients.
Recommandations pratiques
- Il est important d’évoquer le syndrome VEXAS en présence d’une clinique inflammatoire systémique associée à des anomalies hématologiques (myélodysplasie, dyscrasie plasmocytaire, lymphocytose monoclonale B
- Le diagnostic est génétique, basé sur la mise en évidence d’une mutation UBA1 dans les cellules hématopoïétiques par méthode Sanger ou NGS.
- Les traitements les plus prometteurs sont les anticorps monoclonaux anti-IL6 et les inhibiteurs des JAK2. L’allogreffe de cellules souches hématopoïétiques reste actuellement la seule option curative pour les patients jeunes sans comorbidité.
Affiliations
1. Service d’hématologie, Cliniques universitaires Saint-Luc, B-1200 Bruxelles
*Les auteurs ont participé de manière équivalente à l’écriture de l’article.
Correspondance
Dr. Nicole Straetmans
Cliniques Universitaires Saint Luc
Service d’hématologie
Avenue Hippocrate 10, B-1200 Bruxelles
Nicole.straetmans@uclouvain.be
Références
- Beck DB, Ferrada MA, Sikora KA, et al. Somatic Mutations in UBA1 and Severe Adult-Onset Auto-inflammatory Disease. N Engl J Med. 2020;383(27):2628-2638.
- Lytle A, Bagg A. VEXAS: a vivid new syndrome associated with vacuoles in various hematopoietic cells. Blood. 2021;137(26): 3690.
- Grayson P, Patel B, Young N. VEXAS syndrome. Blood. 2021;137(26): 3591-3594.
- Obiorah IE, Patel BA, Groarke EM, et al. Benign and malignant hematologic manifestations in patients with VEXAS syndrome due to somatics mutations in UBA1. Blood advances. 2021;5(16):3202-3215.
- Poulter JA, Collins JC, Cargo C, et al. Novel somatic mutations in UBA1 as a cause of VEXAS syndrome. Blood. 2021;137(26):3676-3681.
- Bourbon E, Heiblig M, Gerfaud Valentin M, et al. Therapeutic options in VEXAS syndrome: insights from a retrospective series. Blood. 2021;137(26):3682-3684.
- Thet Mon O, et al. Thrombosis in VEXAS Syndrome. J ThrombThrombol. 2021; 24;1-6.
- Ferrada M, Savic S, et al. Genotype and Transfusion Dependence Predicts Mortality in VEXAS Syndrome, a Newly Described Disease With Overlap Inflammatory and Hematologic Features. Arthritis Rheumatol. 2021;73(suppl 10).
- Diarra A, Duployez N, Fournier E, et al. Successful allogeneic hematopoietic stem cell transplantation in patients with VEXAS syndrome: a two center experience. Blood Adv. 2021. Published online. DOI : 10.1182/bloodadvances.2021004749.