Cas clinique
Nous présentons le cas d’une patiente de 26 ans primigeste nullipare référée dans notre institution à 26 semaines d’aménorrhées pour échographie référée suite à la mise en évidence à l’échographie de dépistage des malformations fœtales suivantes : retard de formation de la vallée sylvienne, artère ombilicale unique, croissance des os longs au 10ème percentile, septum pellucidum mal visualisé. Le doppler des artères utérines est pathologique bilatéralement avec présence de notch dans un contexte de tabagisme actif.
La patiente ne présente pas d’antécédents médicaux ou chirurgicaux particuliers ni d’antécédents familiaux de malformations congénitales, de retard mental ou de désordre génétique. Les parents sont en bonne santé et ne sont pas consanguins. La patiente n’a pas de maladies infectieuses objectivées pendant la grossesse.
Le test de dépistage prénatal non invasif (NIPT) avec la technique « whole-genome » n’a pas mis en évidence d’aneuploïdies des chromosomes 21, 18 et 13, et le sexe est féminin.
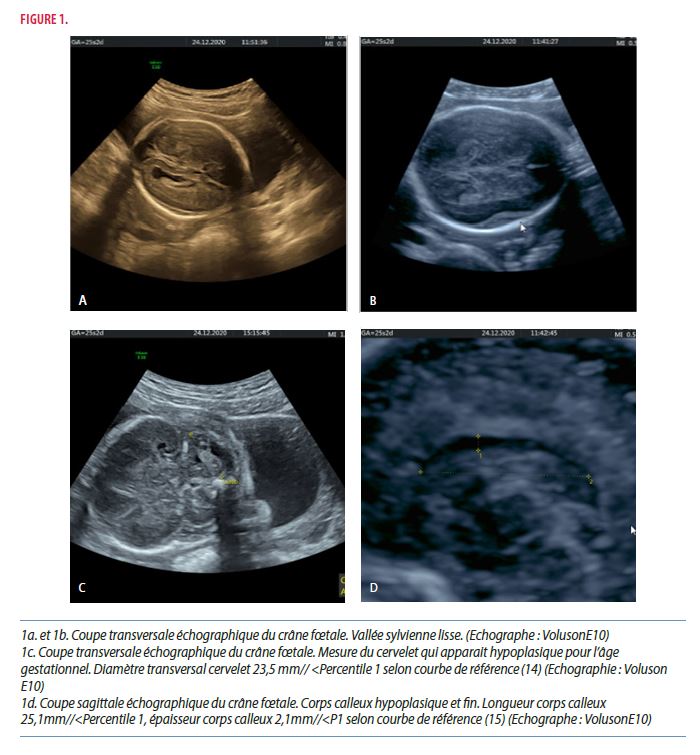
Nous avons effectué une échographie morphologique de référence avec mise en évidence des malformations suivantes au niveau cérébral : retard de gyration (Figure 1a, figure 1b), augmentation des espaces péri-cérébraux, hypoplasie cérébelleuse (Figure 1c) et corps calleux hypoplasique (Figure 1d).
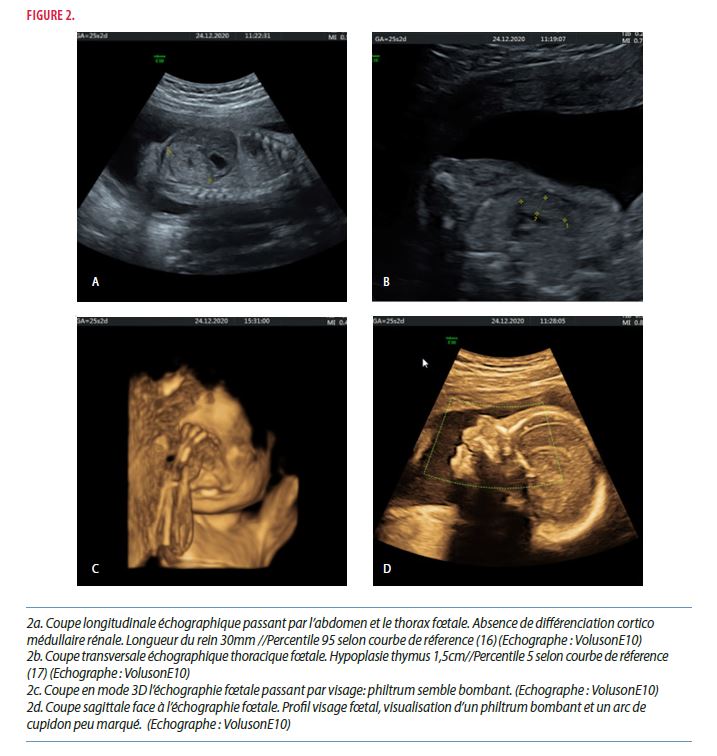
Par ailleurs, une absence de différenciation cortico-médullaire rénale (Figure 2a), une hypoplasie thymique (Figure 2b), une artère ombilicale unique, un retard de croissance intra-utérin et un polyhydramnios ont été mis en évidence au niveau de la morphologie extra-cérébrale. Le facies présente un philtrum bombant et un arc de cupidon peu marqué (Figure 2c et 2d), un retrognatisme avec une lame palatine non visualisée pouvant faire suspecter une fente palatine.
Devant la présence des signes échographiques décrits, la première hypothèse diagnostique évoquée est un syndrome de Di George (délétion 22q11.2).
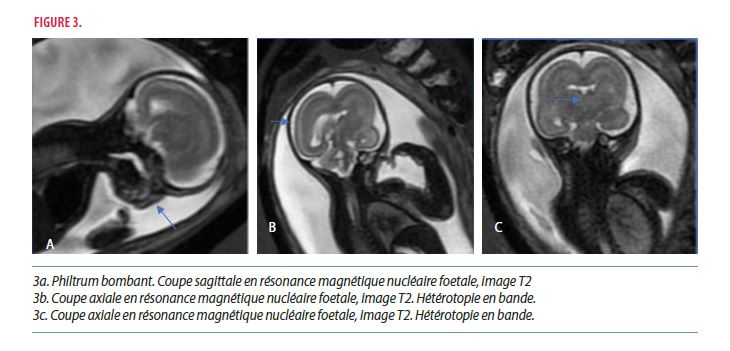
Une résonance magnétique nucléaire (RMN) cérébrale fœtale a été réalisée afin de définir avec précision les malformations cérébrales (Figure 3a, 3b et 3c). Malgré un examen artefacté par une claustrophobie maternelle, un polyhydramnios et des mouvements fœtaux, la RMN a confirmé les signes échographiques avec mise en évidence d’une microcéphalie harmonieuse en sus et sous tentoriel et un cortex fin avec hétérotopie en bande suggérant un arrêt précoce de la migration neuronale. Le corps calleux n’a pas pu être correctement visualisé. L’examen par RMN montre une agyrie qui évoque une lissencéphalie de type 1 et n’est donc pas en faveur d’un syndrome de Di Georges.
Nous avons réalisé une amniocentèse afin de rechercher une cause génétique par FISH (Fluorescence in situ hybridization) et CGH Array (Array comparative genomic hybridization). La recherche des microremaniements par FISH ne révèle pas de microdélétion 22q11.2, permettant d’exclure un syndrome de Di George.
Cependant, la CGH array a mis en évidence une microdélétion de 3,8Mb à l’extrémité télomérique du chromosome 17 (locus 17p13.2-13.3). La microdélétion détectée est un syndrome des gènes contigus, emportant la région minimale critique d’un microremaniement récurrent bien décrit dans la littérature et dans les bases de données de variants pathogènes nous permettant de conclure à un diagnostic de syndrome de Miller-Dieker.
Le diagnostic anténatal a été discuté lors d’une réunion pluridisciplinaire associant obstétriciens, neuropédiatres, rééducateurs fonctionnels, radiologues et généticiens. Le couple a pu rencontrer en consultation les neuropédiatres et les généticiens afin de les aider au mieux pour comprendre le pronostic de l’affection. En raison du pronostic sévère causé par l’absence de gyration et l’association à d’autres malformations caractéristiques du syndrome de Miller-Dieker, le couple a fait une demande d’interruption médicale de grossesse. Celle-ci a été acceptée et réalisé à 30 semaines et 2 jours d’aménorrhée. Un foeticide a été réalisé et la patiente a donné naissance à un enfant mort-né de sexe féminin pesant 1030g.
L’autopsie fœtale a mis en évidence une dysmorphie faciale importante avec une séquence de Pierre Robin (microrétrognatisme, glossoptose, fente palatine postérieure), une artère ombilicale unique et sur le plan neuropathique l’encéphale est caractérisé par une agyrie, un défaut d’operculisation de la vallée sylvienne, un corps calleux mince et partiellement laminé. Histologiquement il existe une inversion du rapport substance grise/substance blanche avec une organisation du cortex en 4 couches en faveur d’une lissencéphalie de type 1.
Discussion
Le développement cortical a lieu en trois étapes superposées : la prolifération cellulaire, la migration neuronale et l’organisation corticale. La migration neuronale se fait entre 6 et 15 semaines d’aménorrhée.
La lissencéphalie est une malformation du développent cortical liée à une anomalie de la migration neuronale vers le cortex, résultant en l’absence de sillons et de gyri. La diminution du nombre de gyri se nomme la pachygirie. La forme la plus sévère est une absence complète de gyration et de sillons (agyrie) en association avec un cortex anormalement épais et mal organisé formé de 4 couches à la place des 6 couches corticales normales (1).
Il existe deux types de lissencéphalie. Celle de type 1 est caractérisée par une agyrie complète. Elle peut être isolée ou associée à des dysmorphies faciales (comprenant le syndrome de Miller-Dieker ou le syndrome de Normal-Roberts). La lissencéphalie de type 2, appelée également “cobblestone” est caractérisée par une désorganisation corticale avec une surface bosselée et granuleuse ainsi qu’un effacement des gyri (par exemple dans le syndrome de Walker Warburg). La lissencéphalie isolée, se distingue du syndrome de Miller-Dieker par l’absence de toute dysmorphie faciale et elle n’est pas associée à une délétion 17p (2).
La lissencéphalie peut être diagnostiquée en échographie anténatale à partir de 24 à 26 semaines d’aménorrhée. Le développement normal du cortex cérébral et son aspect échographique est important à connaitre pour pouvoir visualiser un retard et une absence de développement des sillons. Les structures à analyser sont la scissure pariéto-occipitale, calcarine et sylvienne. L’élément typique à mettre en évidence est l’absence de la scissure sylvienne donnant un aspect anormalement « lisse » au cortex cérébral. Cela peut être diagnostiqué à partir de 24 semaines d’aménorrhée. Un diagnostic de retard de développement cortical avant 20 semaines d’aménorrhée ne peut pas être confirmé car la scissure sylvienne peut encore apparaître lisse avant ce terme et les autres scissures ne sont pas encore développées (3).
D’autres anomalies cérébrales peuvent être associées à une lissencéphalie. C’est le cas de l’agénésie ou la dysplasie du corps calleux et la ventriculomégalie. Par ailleurs, comme décrit par Fong et al., la ventriculomégalie peut être le premier signe visible d’une anomalie de développement cortical et est détectable avant l’anomalie de gyration (3). En cas de suspicion de lissencéphalie, la résonance magnétique nucléaire se révèle être très importante dans le diagnostic précis de l’atteinte cérébrale et également dans la mise au point d’anomalies extra-cérébrales associées (4).
L’imagerie par résonance magnétique nucléaire permet, entre autres, de réaliser le diagnostic différentiel avec le syndrome de Di George, dans lequel les anomalies cérébrales attendues sont une polymicrogyrie, une dysplasie corticale et une hypoplasie cérébelleuse (5).
Le syndrome de Miller-Dieker associe une lissencéphalie sévère à un cortex épaissi. D’autres anomalies cérébrales peuvent être retrouvées, notamment une hypoplasie du corps calleux et/ou du vermis. Des caractéristiques dysmorphiques ainsi que d’autres malformations (cardiaque notamment) peuvent être présentes chez les patients porteurs de ce syndrome. Au niveau de la dysmorphie faciale, les éléments les plus caractéristiques sont un front proéminent, un petit nez, une lèvre supérieure proéminente, des oreilles basses implantées et une petite mâchoire. Les autres malformations majeures fréquemment associées sont une microcéphalie, un retard de croissance intra-utérin, un polyhydramnios, des malformations cardiaques congénitales (dont la tétralogie de Fallot, le défet de septum ventriculaire ou la sténose valvulaire pulmonaire), des malformations gastro-intestinales comme un omphalocèle, des anomalies génito-urinaires, des dysplasies kystiques rénales et des fentes palatines (6).
Le polyhydramnios est probablement lié à l’atteinte sévère du système nerveux central engendrant une difficulté de déglutition chez le fœtus (7).
Chih-Ping Chen et al., ont rapporté dans une revue de littérature, les malformations échographiques les plus fréquentes retrouvées dans 29 diagnostics anténataux de syndrome de Miller-Dieker : un polyhydramnios dans 66% des cas, un retard de croissance intra utérin dans 62% des cas, une ventriculomégalie dans 59% des cas, une agénésie ou dysgénésie du corps calleux dans 41% des cas, une malformation cardiaque congénitale dans 14% des cas, et enfin, un omphalocèle et des anomalies rénales dans 7% des cas (8). C’est l’association avec des malformations congénitales cardiaques de type conotroncale qui pose le diagnostic différentiel de syndrome de Di George.
La mise en évidence à l’échographie fœtale prénatale d’une agyrie associée à un retard de croissance et un polyhydramnios nécessite une mise au point génétique. Une délétion du locus 13.3 sur le chromosome 17 (gène LIS1) peut être mise en évidence par FISH, posant le diagnostic du syndrome de Miller-Dieker (9).
Approximativement 80% des syndromes de Miller-Dieker sont liés à une microdélétion de novo. Pour les 20% restants, l’origine de la délétion provient d’un réarrangement chromosomique parental équilibré (10). Il est donc nécessaire de réaliser une analyse génétique parentale (caryotype et FISH 17p13.3) et de rechercher des réarrangements chromosomiques afin de préciser le conseil génétique avec proposition de diagnostic préimplantatoire dans l’éventualité d’une future grossesse (11).
Le test de dépistage prénatal non invasif (NIPT) est utilisé pour le dépistage fœtal des trisomies les plus fréquentes (21, 13 et 18). Il a été démontré que le NIPT avec séquen-çage « whole-genoma » peut également détecter des délétions et duplications chromosomiques. Néanmoins, le test est confronté à certaines limites : la détection d’anomalies chromosomiques dont la signification clinique est inconnue, un nombre élevé de faux positifs et la difficulté de détecter des anomalies de petite taille (moins de 5Mb). Les délétions et duplications présentes dans la majorité des syndromes (comme le syndrome de Miller-Dieker) font moins de 5Mb. L’utilisation du NIPT pour le dépistage de syndromes causés par des microdélétions/microduplications peut avoir un effet bénéfique pour la prise en charge de grossesses à risque. Étant donné que la majorité des délétions chromosomiques pathogènes surviennent de novo, l’utilisation du NIPT est également utile dans la population générale non à risque (12).
La sévérité et le pronostic sont déterminés par le degré de lissencéphalie c’est-à-dire le degré de développent des sillons. En cas d’agyrie, le pronostic est réservé, d’où l’importance du diagnostic anténatal pour pouvoir orienter la prise en charge obstétricale. L’étude De Rijk (13) a montré qu’en moyenne, l’âge de décès des enfants atteints par le syndrome de Miller-Dieker est de 3,5 ans, le plus souvent dû à une infection ou une épilepsie.
Le développement incomplet du cerveau provoque un retard de développement majeur, une déficience intellectuelle, une hypotonie, des crises épileptiques fréquentes et des manifestations neurologiques variables mais souvent sévères, comme des troubles alimentaires importants. Les crises épileptiques liées à une encéphalopathie épileptogène surviennent chez 90% des enfants avant l’âge de 6 mois. La mort de ces enfants a lieu tôt dans l’enfance, le plus souvent avant l’âge de deux ans (3). La cause la plus fréquente est le mauvais contrôle des voies respiratoires, causant une pneumonie d’inhalation (9).
Afin d’être en mesure de poser un diagnostic échographique prénatal, il est essentiel d’être familiarisé avec l’apparence normale du cortex cérébrale aux différents stades de la maturation fœtale. Compte tenu de l’espérance de vie réduite des nouveau-nés atteints (moins de deux ans dans la majorité des cas), le diagnostic prénatal rend possible une discussion éclairée « parents-médecins », afin d’opter pour une prise en charge obstétricale adéquate et adaptée aux souhaits du couple.
Conclusion
Nous avons présenté un cas de syndrome de Miller-Dieker diagnostiqué en anténatal avec comme premier signe la mise en évidence échographique d’une agyrie et d’un retard de croissance intra-utérin et d’une dysmorphie faciale avec retrognatisme et possible séquence de Pierre Robin.
La résonance magnétique nucléaire a permis de préciser l’anomalie cérébrale et le diagnostic définitif a été posé par l’analyse génétique. Le principal diagnostic différentiel a été posé avec le syndrome de Di George, car la présence d’une hypoplasie cérébelleuse avec ventriculomégalie associée à un retard de croissance intra-utérin, polyhydramnios, dysmorphie faciale et hypoplasie thymique sont des signes caractéristiques de la délétion 22q11.
En illustrant ce cas nous voudrions accentuer l’importance de l’échographie de dépistage qui est le moyen diagnostic le plus accessible et utile pour suspecter le diagnostic durant le 3ème ou 4ème mois de grossesse : certaines scissures cérébrales sont donc importantes à visualiser comme la scissure parieto-occipitale et calcarine qui sont facilement évaluables en coupe transversale du cerveau.
Recommandations pratiques
La mise en évidence d’une dysmorphie faciale de type séquence de Pierre Robin à l’échographie associée à un retard de croissance intra-utérin et une ventriculomégalie nécessite un suivi échographique foetale et une résonance magnétique nucléaire pour préciser l’atteinte cérébrale et rechercher une lissencéphalie. Le type de lissencéphalie mis en évidence permet d’affiner la recherche génétique, en concertation avec les généticiens, pour permettre d’atteindre un diagnostic.
Conflit d’intérêt
Les auteurs déclarent n’avoir aucun conflit d’intérêt
Affiliations
Département d’obstétrique, Centre Hospitalier Luxembourg (CHL), Luxembourg
Correspondance
Dr. Marta Merola Martinez
Centre Hospitalier du Luxembourg
Service de Gynécologie-Obstétrique
marta.merola@gmail.com
Références
- McGahan JP, Grix A, Gerscovich EO. Prenatal diagnosis of lissencephaly : Miller-Dieker syndrome. Clin Utrasound. 1994 ; 22 :560-563.
- Batanian JR. et al. Rapid diagnosis of Miller-Dieker syndrome and isolated lissencephaly sequence by the polymerase chain reaction. Human Genetics. 1990; 85:555–559.
- Fong KW, Ghai S, Toi A, Blaser S, Winsor EJ, Chitayat D. Prenatal ultrasound findings of lissencephaly associated with Miller-Dieker syndrome and comparison with pre-and postnatal magnetic resonance imaging. Ultrasound Obstet Gynecol. 2004 ; 24 :716-723.
- Ghai S, Fong KW, Toi A, Chitayat D, Pantazi S, Blaser S. Prenatal US and MR Imaging Findings of Lissencephaly: Review of Fetal Cerebral Sulcal Development. RadioGraphics. 2006 26:2, 389-405.
- Bohm LA, Zhou TC, Mingo TJ, Dugan SL, Patterson RJ, Sidman JD, Roby BB. Neuroradiographic findings in 22q11.2 deletion syndrome. Am J Med Genet A. 2017 Aug; 173(8):2158-2165.
- Chen CP, Liu YP, Lin SP, Chen M, Tsai FJ, Chen YT, et al. Ventriculomegaly, intrauterine growth restriction, and congenital heart defects as salient prenatal sonographic findings of Miller-Dieker lissencephaly syndrome associated with monosomy 17p in a fetus. Taiwan J Obstet Gynecol. March 2010 Mar;49(1):81-6.
- McGahan JP, Grix A, Gerscovich EO. Prenatal diagnosis of Lissencephaly : Miller-Dieker syndrome. J Clin Ultrasound. 1994; 22:560-563.
- Chen CP, Chang TY, Guo WY, Wu PC, Wang LK, Chern SR, et al. Chromosome 17p13.3 deletion syndrome: a CGH characterization, prenatal findigs and diagnosis, and literature review. Gene. 2013; 532:152-159.
- Lenzini E, D’Ottavio G, Città A, Gambel Benussi D, Petix V, Pecile V. Prenatal diagnosis of Miller-Dieker syndrome by ultrasound and molecular cytogenetic analysis. Clin Genet. 2007; 72:487-489.
- Stratton RF, Dobyns WB, Airhart SD, Ledbetter DH. New chromosomal syndrome: Miller-Dieker syndrome and monosomy 17p13. Hum Genet. 1984;67(2):193-200.
- William B Dobyns, Soma Das. PAFAH1B1-Associated Lissenceohaly/Subcortical Band Heterotopia. Gene Reviews. 2014.
- Neofytou MC, Tsangaras K, Kypri E, Loizides C, Ioannides M, Achilleos A, et al. Targeted capture enrichment assay for non-invasive prenatal testing of large and small size sub-chromosomal deletions and duplications. PLoS One. 2017 Feb 3;12(2):e0171319.
- De Rijk-van Andel JF, Arts WF, Hofman A, Staal A, Niermeijer MF. Epidemiology of lissencephaly type I. Neuroepidemiology. 1991;10(4):200-4.
- Chavez MR, Ananth CV, Smulian JC, Lashley S, Kontopoulos EV, Vintzileos AM. Fetal transcerebellar diameter nomogram in singleton gestations with special emphasis in the third trimester: a comparison with previously published nomograms. Am J Obstet Gynecol. 2003 Oct;189(4):1021-5.
- Harreld JH, Bhore R, Chason DP, Twickler DM. Corpus callosum length by gestational age as evaluated by fetal MR imaging. AJNR Am J Neuroradiol. 2011;32(3):490-494.
- Van Vuuren SH, Damen-Elias HA, Stigter RH, van der Doef R, Goldschmeding R, de Jong TP, et al. Size and volume charts of fetal kidney, renal pelvis and adrenal gland. Ultrasound Obstet Gynecol. 2012 Dec;40(6):659-64.
- Cho JY, Min JY, Lee YH, McCrindle B, Hornberger LK, Yoo SJ. Diameter of normal fetal thymus on ultrasound. Ultrasound Obstet Gynecol. 2007 Jun ;29(6) :634-8.