Cas clinique
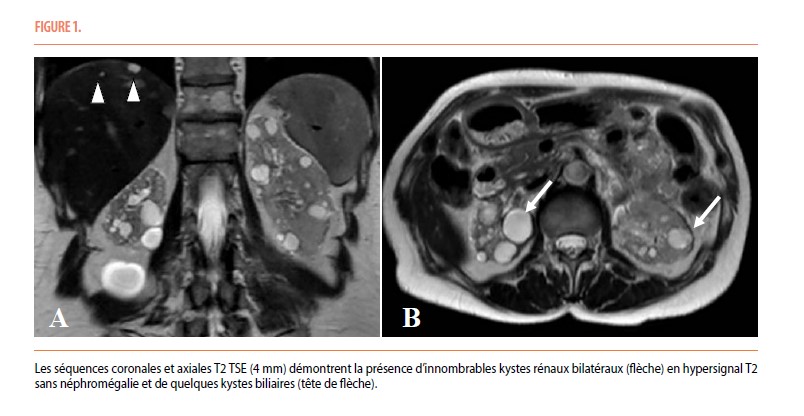
Nous rapportons le cas d’une patiente de 64 ans qui s’est présentée en consultation de néphrologie en 2013 pour l’évaluation d’une maladie rénale chronique associée à une hypertension artérielle. Le bilan a révélé une créatinine sérique à 2 mg/dl, soit un débit de filtration glomérulaire estimé (DFGe) à 27 ml/min/1,73 m², un rapport protéinurie/créatininurie à 340 mg/g (N < 150 mg/g) et l’absence d’hématurie. L’échographie abdominale a montré des reins de 105 et 115 mm de longueur et la présence de multiples kystes rénaux bilatéraux. L’IRM a ensuite confirmé la présence d’innombrables kystes rénaux bilatéraux associés à quelques kystes biliaires centimétriques (Figure 1). Les volumes des reins droit et gauche étaient mesurés à 115 ml et 148 ml respectivement, soit normaux.
À l’anamnèse, il n’y avait pas d’antécédent familial de polykystose rénale ou de maladie rénale chronique. La fille cadette de la patiente avait été opérée d’une fente labiale, était appareillée pour une surdité neurosensorielle et présentait un retard de développement psychomoteur, survenu dans le décours d’une infection congénitale à CMV. À l’âge de 22 ans, elle avait une fonction rénale normale, et l’échographie révélait trois kystes rénaux corticaux à gauche. La fille aînée de la patiente présentait une fonction rénale normale et l’absence de kystes rénaux à l’échographie à l’âge de 29 ans.
Une analyse génétique a été réalisée pour la première fois chez la patiente en 2014. Le séquençage Sanger et l’analyse MLPA (Multiplex Ligation-dependent Probe Amplification) des gènes PKD1, PKD2, HNF1B et UMOD n’a pas révélé de variant causal. En 2022, une nouvelle analyse génétique à base d’un panel de gènes impliqués dans les ciliopathies a révélé un variant causal c.935+5_935+8del du gène OFD1. Ce variant entraine une altération de l’épissage de la protéine. Une anamnèse complémentaire de notre patiente a révélé des antécédents personnels de correction chirurgicale d’une fente palatine et d’une grossesse avec mort in utero à 5 mois d’un fœtus masculin, dans le contexte de malformations multiples (cardiaque et rénale avec présence de polydactylie). La patiente a atteint le stade d’insuffisance rénale terminale à l’âge de 64 ans et débuté la dialyse péritonéale.
Discussion
Le syndrome oro-facio-digital de type I (OFD1) est une maladie héréditaire transmise par le chromosome X. Sa prévalence est de 1/50000 à 1/250000. Le syndrome OFD1 se présente de manière sporadique (sans histoire familiale) dans 75% des cas et est responsable d’une mortalité prénatale chez les fœtus masculins, comme dans le cas rapporté (1). Les manifestations cliniques de l’OFD1 sont orales (langue lobulée, nodules linguaux, fente palatine, anomalies dentaires), faciales (hypertélorisme, arête nasale élargie, hypoplasie des ailes du nez, micrognathie) et digitales (brachydactylie, syndactylie, clinodactylie et polydactylie) (1). En outre, 40% des patients peuvent présenter des malformations du système nerveux central et une déficience intellectuelle légère à modérée. Des reins plurikystiques sont décrits dans 50% des cas, non retrouvés dans l’enfance et plus fréquemment associés à des variants affectant l’épissage d’OFD1 comme celui observé chez la patiente (2,3). Dans une étude rétrospective portant sur 36 patients, 82% des patients OFD1 avec polykystose rénale ont évolué vers une maladie rénale chronique terminale à un âge médian de 36 ans (4). Les patients avec syndrome OFD1 bénéficient similairement des techniques de suppléance rénale, dont la greffe de rein, en comparaison aux patients avec d’autres néphropathies initiales (5).
Le syndrome OFD1 fait partie de la grande famille des ciliopathies, maladies génétiques ayant en commun un dysfonctionnement des cils primaires. Ceux-ci sont des organites cellulaires spécialisés agissant comme antenne à la surface de nombreuses cellules de l’organisme (6). Plus de 35 ciliopathies sont connues. Les ciliopathies présentent des phénotypes similaires dont maladie rénale kystique, malformations cérébrales, fibrose hépatique, anomalies rétiniennes et du squelette. Le lien physiopathologique entre les mutations des protéines du cil primaire, le défaut de structure et de fonction du cil et la formation et croissance de kystes à partir de l’épithélium tubulaire rénal est encore mal compris (6).
Le diagnostic différentiel des plurikystoses comporte d’autre ciliopathies ainsi que d’autres maladies acquises et héréditaires (Tableau). Avant de détailler succinctement les maladies kystiques héréditaires, nous rappelons la présence fréquente de kystes rénaux simples dans la population générale. Plus de la moitié des personnes âgées de >50 ans ont au moins un kyste rénal. De même, les kystes rénaux acquis sont plus fréquents chez les patients porteurs d’une maladie rénale chronique avancée, en particulier les patients dialysés (7) (Tableau). La classification de Bosniak est utilisée pour stratifier le risque de malignité des kystes rénaux. Cette classification est basée sur les caractéristiques d’imagerie des kystes rénaux (aspect des parois, contenu et densité du kyste, présence de septa, calcifications), principalement obtenues sur base de la tomodensitométrie (8).
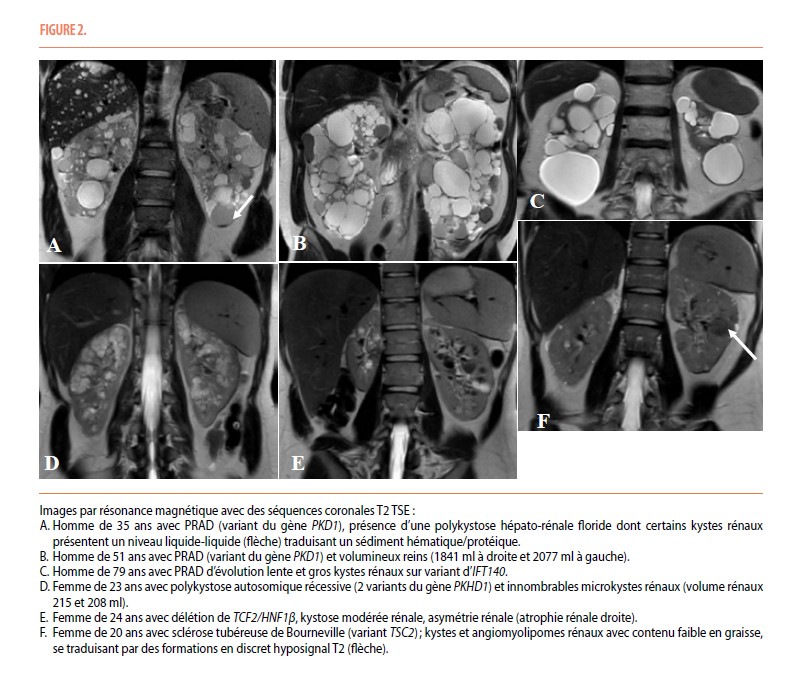
Parmi les maladies kystiques héréditaires, la polykystose rénale autosomique dominante (PRAD ou ADPKD en anglais), autre ciliopathie, est la plus fréquente, avec une prévalence d’environ 1/1000-1/2500. La PRAD est caractérisée par la formation et la croissance continue de kystes rénaux, entrainant une augmentation de la taille des reins parallèlement à une réduction du DFGe (9,10) (Tableau, Figure 2). La kystose rénale est de taille variable. L’hypertension est souvent précoce et la plupart des patients présentent des kystes hépatiques, le plus souvent asymptomatiques. Les anévrysmes cérébraux sont présents chez 4-10% des patients, notamment en cas d’histoire familiale d’anévrysme ou d’hémorragie cérébrale (9,10). Une histoire familiale de PRAD est retrouvée dans 85-90% des cas. Bien que les variants pathogènes dans les gènes PKD1 et PKD2 soient associées à >90% des cas de PRAD, d’autres gènes causaux ont récemment été décrits (dont les gènes GANAB, DNAJB11, IFT140, ALG5, ALG8, ALG9 et NEK8) (11-14). Les gènes PKD1 et PKD2 codent pour la polycystine 1 et la polycystine 2 respectivement, deux protéines qui forment un complexe présent au niveau du cil primaire des cellules tubulaires rénales (6) (Figure 2). Il faut noter que la PRAD liée aux gènes non-PKD1-2 est souvent caractérisée par une kystose rénale moins développée, une kystose hépatique variable et une réduction moins rapide du DFGe. Le diagnostic de PRAD se fait sur base de l’histoire familiale et du nombre de kystes rénaux à l’échographie et/ou à l’IRM en fonction de l’âge (15). Un test génétique peut aussi confirmer la présence du variant causal connu chez un autre membre de la famille atteint de PRAD ou détecter le variant causal chez le propositus. La moitié des patients avec PRAD arriveront au stade de la maladie rénale chronique terminale à l’âge de 60 ans (9). Le Tolvaptan (Jinarc®), un antagoniste sélectif du récepteur V2 de la vasopressine au niveau des cellules tubulaires rénales, est remboursé en Belgique depuis 2016 pour le traitement des patients porteurs d’une PRAD à risque de progression rapide (16).
Les autres ciliopathies associées à une plurikystose rénale sont la polykystose rénale autosomique récessive et les néphronophtises. Bien que se manifestant généralement dans l’enfance, la polykystose rénale autosomique récessive peut parfois se présenter à l’âge adulte. Les variants pathogènes du gène PKHD1 sont responsables de la majorité des cas. La fibrocystine, codée par ce gène, se trouve au niveau des tubules distaux des reins et des canaux biliaires du foie. La polykystose rénale autosomique récessive est fréquemment associée à une fibrose hépatique congénitale et des microkystes rénaux (17).
Les néphronophtises sont transmises de manière autosomique récessive et sont associées à des gènes encodant différentes protéines du cil primaire. Elles sont caractérisées par la présence de kystes cortico-médullaires sans majoration du volume rénal et des manifestations extra-rénales telles qu’une rétinite pigmentaire et une fibrose hépatique. Plus de 25 gènes causaux sont connus dont le gène NPHP1 qui est le plus fréquemment impliqué (6,18).
En dehors des ciliopathies, d’autres maladies héréditaires sont associées à la plurikystose rénale. Les néphropathies tubulo-interstitielles autosomiques dominantes (autosomal dominant tubulo-interstitial kidney disease, ADTKD) peuvent entrainer une kystose rénale peu importante, sans néphromégalie. Ces maladies sont associées à des variants pathogènes dans différents gènes, dont MUC1, UMOD et TCF2/HNF1B (19).
La sclérose tubéreuse de Bourneville, de transmission autosomique dominante, est caractérisée par la présence d’hamartomes multiples systémiques notamment au niveau cutané, rénal, pulmonaire, du système nerveux central et osseux. Les manifestations rénales comportent les kystes, les angiomyolipomes et les carcinomes (20). Des variants pathogènes dans les gènes TSC1-2 sont responsables du phénotype de sclérose tubéreuse de Bourneville. Il existe une entité rare de syndrome des gènes contigus TSC2/PKD1 résultant de la délétion des 2 gènes cités et entrainant un phénotype hybride entre la sclérose tubéreuse et la PRAD.
La maladie de Von Hippel Lindau est une autre maladie rare prédisposant au développement de tumeurs bénignes et malignes, due à des variants pathogènes de VHL. Elle est caractérisée par la présence d’hémangioblastomes du système nerveux central et de la rétine, de phéochromocytomes, de kystes pancréatiques et de kystes et carcinomes rénaux (21).
Les progrès constants dans le domaine des méthodes de séquençage à haut débit et la baisse des coûts associés ont amélioré le rendement diagnostique et ont facilité l’accessibilité à ces tests génétiques dans la pratique clinique. Les critères motivant une analyse génétique devant une plurikystose incluent la présence d’une histoire familiale de néphropathie et/ou maladie rénale kystique, une kystose importante, une néphromégalie et la présence d’atteinte(s) extra-rénale(s) suggestive(s) d’une maladie héréditaire. Le recours à une analyse de panel de gènes (comportant un grand nombre de gènes potentiellement impliqués dans la kystose rénale dont PKD1, PKD2 et les autres gènes repris dans Tableau) ou un séquençage de l’exome peut être très utile à titre diagnostique et pronostique. Le résultat génétique peut servir au conseil génétique (notamment risque de transmission aux enfants et possibilité de recours au diagnostic préimplantatoire), à identifier les maladies kystiques pour lesquelles un traitement spécifique est disponible (tolvaptan dans la PRAD, everolimus dans la sclérose tubéreuse de Bourneville) et à chercher les manifestations extra-rénales potentiellement associées (22) (Tableau).
Il est aussi intéressant de noter que l’anamnèse complémentaire chez notre patiente a permis de mettre en évidence des caractéristiques associées à sa maladie génétique (correction d’une fente palatine et mort in utero d’un fœtus masculin). Ceci est un phénotypage inversé, qui consiste à caractériser les phénotypes associés après avoir identifié le variant génétique causal. Cette approche est utilisée en génétique et recherche biomédicale pour étudier les maladies génétiques rares ou complexes.
Conclusions
Le diagnostic différentiel de la polykystose rénale comporte plusieurs maladies rénales génétiques dont les ciliopathies. La pose du diagnostic nécessite une approche multidisciplinaire clinique, radiologique et génétique afin de pouvoir appréhender au mieux le pronostic, la prise en charge rénale et extra-rénale et les implications génétiques spécifiques de chaque maladie. Notre cas illustre l’importance d’une anamnèse personnelle et familiale détaillée et l’aide diagnostique significative des nouvelles techniques de séquençage génétique.
Références
- Gurrieri F, Franco B, Toriello H, Neri G. Oral–Facial–Digital Syndromes: Review and Diagnostic Guidelines. Am J Med Genet. 2007;143A(24):3314-3323. doi:10.1002/ajmg.a.32019.
- Thauvin-Robinet C, Cossée M, Cormier-Daire V, Van Maldergem L, Toutain A, Alembik Y, et al. Clinical, molecular, and genotype-phenotype correlation studies from 25 cases of oral-facial-digital syndrome type 1: a French and Belgian collaborative study. J Med Genet. 2006;43(1):54-61. doi: 10.1136/jmg.2004.027672.
- Brunella F, Thauvin-Robinet C. Update on oral-facial-digital syndromes (OFDS). Cilia 2016;5 :12. doi :10.1186/s13630-016-0034-4.
- Saal S, Faivre L, Aral B, Gigot N, Toutain A, Van Maldergem L, et al. Renal insufficiency, a frequent complication with age in oral-facial-digital syndrome type I. Clin Genet. 2010;77(3):258-65. Doi: 10.1111/j.1399-0004.2009.01290.x.
- Halleux D, Dahan K, Thauvin-Robinet C, Aral B, Pouteil-Noble C, Devuyst O, et al. Outcome of kidney transplantation in type I oral-facial-digital syndrome. Transpl Int. 2011; 24(6):e48-50. Doi:10.1111/j.1432-éé77.2010.01208.x.
- McConnachie DJ, Stow JL, Mallett AJ. Ciliopathies and the Kidney: A Review. Am J Kidney Dis. 2021;77(3):410-419. doi:10.1053/j.ajkd.2020.08.012.
- Eknoyan G. A clinical view of simple and complex renal cysts. J Am Soc Nephrol. 2009;20(9):1874-1876. doi:10.1681/ASN.2008040441.
- Silverman SG, Pedrosa I, Ellis JH, Hindman NM, Schieda N, Smith AD, et al. Bosniak Classification of Cystic Renal Masses, Version 2019: An Update Proposal and Needs Assessment. Radiology. 2019;292(2):475-488. Doi:10.1148/radiol.2019182646.
- Chebib FT, Torres VE. Autosomal Dominant Polycystic Kidney Disease: Core Curriculum 2016. Am J Kidney Dis. 2016;67(5):792-810. doi:10.1053/j.ajkd.2015.07.037.
- Cornec-Le-Gall E, Alam A, Perrone RD. Autosomal dominant polycystic kidney disease. Lancet. 2019; 393: 919-935. doi:10.1016/S0140-6736(18)32782-X.
- Cornec-Le Gall E, Torres VE, Harris PC. Genetic Complexity of Autosomal Dominant Polycystic Kidney and Liver Diseases. J Am Soc Nephrol. 2018;29(1):13-23. doi:10.1681/ASN.2017050483.
- Lanktree MB, Haghighi A, di Bari I, Song X, Pei Y. Insights into Autosomal Dominant Polycystic Kidney Disease from Genetic Studies. Clin J Am Soc Nephrol. 2021;16(5):790-799. doi:10.2215/CJN.02320220.
- Senum SR, Li YSM, Benson KA, Joli H, Olinger E, Lavu S, et al. Monoallelic IFT140 pathogenic variants are an important cause of the autosomal polycystic kidney-spectrum phenotype. Am J Hum Genet. 2022;109(1):136-156. doi:10.1016/j.ajhg.2021.11.016.
- Claus LR, Chen C, Stallworth J, Turner JL, Slaats GG, Hawks AL, et al. Certain heterozygous variants in the kinase domain of the serine/threonine kinase NEK8 can cause an autosomal dominant form of polycystic kidney disease. Kidney Int. 2023;104(5):995-1007. doi:10.1016/j.kint.2023.07.021.
- Pei Y, Hwang YH, Conklin J, Sundsbak JL, Heyer CM, Chan W, et al. Imaging-based diagnosis of autosomal dominant polycystic kidney disease. J Am Soc Nephrol. 2015;26(3):746-753. doi:10.1681/ASN.2014030296.
- Demoulin N, Ho A, Godefroid N, Clerckx C, Annet L, Pirson Y, et al. Polykystose rénale autosomique dominante – enfin un traitement freinant sa progression! Louvain Med. 2017;136(2):103-109.
- Bergmann C, Guay-Woodford LM, Harris PC, Horie S, Peters DJM, Torres VE. Polycystic kidney disease. Nat Rev Dis Primers. 2018;4(1):50. doi:10.1038/s41572-018-0047-y.
- Luo F, Tao YH. Nephronophthisis: A review of genotype-phenotype correlation. Nephrology (Carlton). 2018;23(11):904-911. doi:10.1111/nep.13393
- Eckardt KU, Alper SL, Antignac C, Bleyer AJ, Chauveau D, Dahan K, et al. Autosomal dominant tubulointerstial disease: diagnosis, classification, and management – A KDIGO consensus report. Kidney Int. 2015;88(4):676-83. doi:10.1038/ki.2015.28.
- Mekahli D, Muller RU, Marlais M, Wlodkowski T, Haberle S, Lopez de Argumedo, et al. Clinical practice recommendations for kidney involvement in tuberous sclerosis complex: a consensus statement by the ERKNet Working Group for Autosomal Dominant Structural Kidney Disorders and the ERA Genes & Kidney Working Group. Nat Rev Nephrol. 2024;20(6):402-420. Doi.10.1038/s41581-024-00818-0.
- Binderup MLM, Smerdel M, Borgwadt L, Nielsen SSB, Madsen MG, Moller HU, et al. von Hippel-Lindau disease: Updated guideline for diagnosis and surveillance. Eur J Med Genet. 2022;65(8):104538. doi.org/10.1016/j.egmg.2022.104538.
- Kottgen A, Cornec-Le Gall E, Halbritter J, Kiryluk K, Mallette AJ, Parekh RS, et al. Genetics in chronic kidney: conclusions from a kidney disease: Improving Global Outcomes (KDIGO) Controversies Conference. Kidney Int. 2022;101(6):1126-1141. Doi.org/10.1016/j.kint.2022.03/019.
Affiliations
1. Néphrologie, Cliniques universitaires Saint-Luc, Brussels, B-1200 Bruxelles, Belgique
2. Institut de Recherche Expérimentale et Clinique, UCLouvain, B-1200 Bruxelles, Belgique
3. Département de Radiologie, Cliniques universitaires Saint-Luc, B-1200 Bruxelles, Belgique
4. Centre de Génétique Humaine, Institut de Pathologie et de Génétique, Gosselies, Belgique
Correspondance
Dre Catherine Verhellen
Cliniques universitaires Saint-Luc
Néphrologie
Avenue Hippocrate 10
B-1200 Bruxelles