Causes systémiques de nécroses cutanées
Cas clinique n°1
Il s’agit d’une patiente de 58 ans connue pour une insuffisance rénale chronique sur néphropathie inconnue, dialysée depuis 2008. Cette patiente, avec antécédent de cardiopathie ischémique et valvulaire, souffre d’hypertension artérielle et d’obésité mais est non diabétique. À l’examen clinique, on retrouve des plaques nécrotiques douloureuses d’extension rapide aux pourtours livédoïdes violacés sur les membres inférieurs et en sous-mammaire droit (Figure 1). Les pouls périphériques sont tous perçus et les pieds sont chauds. Elle est par ailleurs apyrétique.
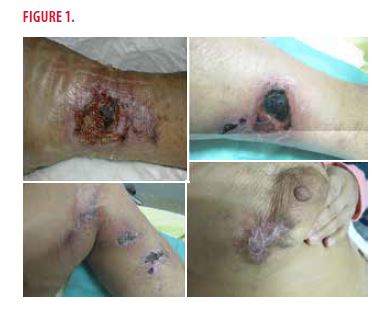
Quels sont les diagnostics à évoquer devant un tel tableau clinique ?
1.Il faut penser à des ulcères d’origine artérielle vu le terrain cardio-vasculaire, le caractère douloureux des ulcères et leur extension rapide. La localisation sur le sein et les pourtours livédoïdes sont par contre totalement atypiques d’ulcères artériels. En outre, les pouls périphériques sont bien perçus et les pieds sont chauds, ce qui va à l’encontre de ce diagnostic chez cette patiente.
2.L’angiodermite nécrotique (ou ulcère de Martorell) est caractérisée par l’apparition d’un ulcère douloureux avec nécrose centrale entourée d’une bordure livédoïde typiquement localisé au niveau de la face antéro-latérale de la jambe. Elle survient surtout chez la femme d’âge mûr et est fréquemment associée à l’hypertension artérielle et au diabète. Ce diagnostic est à évoquer devant ce tableau clinique, car il s’agit d’une femme âgée de plus de 50 ans avec hypertension artérielle présentant des ulcérations aux pourtours livédoïdes. Par contre, les localisations multiples et notamment la localisation au niveau du sein sont complètement atypiques pour une angiodermite nécrotique.
3.Les vascularites nécrosantes comme la périartérite noueuse, la granulomateuse éosinophilique avec polyangéite (anciennement Syndrome de Churg-Strauss) et la granulomatose avec polyangéite (anciennement maladie de Wegener). En général dans la vascularite nécrosante, on observe des lésions de type purpura nécrotique (placard érythémateux infiltré douloureux), un livédo et des ulcérations notamment digitales. Ici, le diagnostic de vascularite nécrosante doit être exclu, car les lésions sont trop uniformes à type de plaques nécrotiques.
4.Finalement, il faut évoquer le diagnostic de calciphylaxie ou artériolopathie urémique calcifiante, car il s’agit d’une patiente obèse, en insuffisance rénale chronique dialysée et qui a probablement une hyperparathyroïdie secondaire.
À la biopsie cutanée, le diagnostic de calciphylaxie est confirmé.
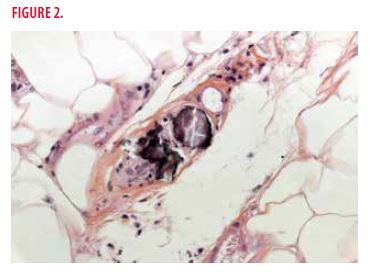
L’analyse histologique montre une calcification circonférentielle (mise en évidence par la coloration Von Kossa) des artérioles hypodermiques associée à une hyperplasie intimale secondaire, une adiponécrose ainsi que des dépôts calciques extravasculaires (Figure 2).
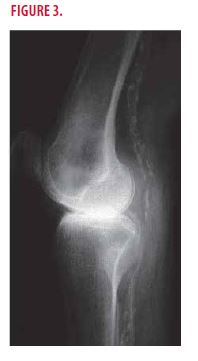
Dans le bilan biologique, on note une hyperparathyroïdie secondaire (PTH= 407 pg/ml ; N<74) et une augmentation du produit phosphocalcique (6,45 mmol2/L2 ; N<4.5). Vu la fréquente association de thromboses dans un contexte de calciphylaxie, une radiographie standard et un angioscanner des membres inférieurs sont réalisés chez cette patiente. L’imagerie montre une infiltration calcique iliaque externe avec des sténoses multiples du tiers moyen de la fémorale superficielle témoignant d’une médiacalcose. On observe également des calcifications sous-cutanées (Figure 3).
Qu’est-ce que la calciphylaxie ?
La calciphylaxie est une complication rare (1% des patients dialysés) mais grave. Elle peut toutefois bien qu’exceptionnellement aussi apparaître en dehors de la dialyse chez des sujets sans insuffisance rénale. La mortalité à 1 an est de 45%. Le terrain typique est formé par la triade : dialysé, femme, obésité.
Cliniquement, la calciphylaxie donne des ulcérations et placards nécrotiques douloureux, indurés, qui siègent soit au niveau proximal, en particulier au niveau des zones avec couche graisseuse importante, soit au niveau distal touchant les extrémités. Les lésions ont également un tropisme pour les zones traumatisées tel que les sites de biopsie et sites d’injection d’héparine.
La physiopathologie reste partiellement inconnue avec des calcifications vasculaires touchant la microcirculation. Au niveau moléculaire, un défaut d’activation de la protéine matricielle GLA, vitamine K dépendante (qui serait protectrice face aux calcifications tissulaires), a été mis en évidence récemment d’où la contre-indication des anti-vitaminiques K dans la calciphylaxie.
Le traitement consiste en l’augmentation des séances de dialyse avec des bains pauvres en calcium et une limitation des apports en vitamine D. Les anti-vitaminiques K doivent être arrêtés et contre-indiqués. Il faut également traiter l’hyperparathyroïdie secondaire soit par parathyroïdectomie soit par cinacalcet (Mimpara®). Le cinacalcet est un calcimimétique. Son mécanisme d’action consiste à diminuer les concentrations plasmatiques de la parathormone (PTH) et du calcium en augmentant la sensibilité au calcium extracellulaire des récepteurs calciques au niveau de la parathyroïde. Finalement, il est recommandé d’administrer du thiosulfate de sodium 5-25g en intraveineux 3x/semaine lors des séances de dialyse et de continuer jusqu’à deux mois après cicatrisation. Les soins locaux des lésions cutanées sont également très importants.
Cas clinique n°2
Il s’agit d’un homme de 50 ans avec hépatite C chronique non réplicative hospitalisé en néphrologie pour une insuffisance rénale aiguë, une neuropathie périphérique sensitivomotrice des quatre membres et des nécroses digitales multiples d’apparition récente (Figure 4).
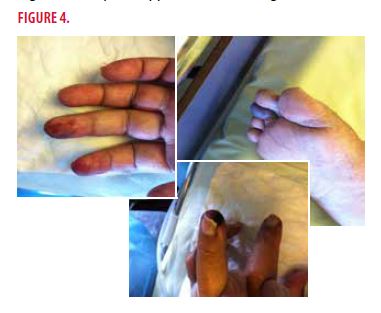
Quels sont les diagnostics à évoquer devant un tel tableau clinique ?
1.Il faut évoquer le diagnostic d’emboles de cholestérol, car il s’agit d’un homme de 50 ans avec des lésions d’aspect livédoïde et une atteinte rénale associée. Par contre, il n’a pas de facteur de risque cardio-vasculaire et surtout il n’y a pas de facteur déclenchant, en particulier chirurgie de l’aorte, cathétérisme artériel et mise sous anticoagulant. De plus, en cas d’emboles de cholestérol, l’atteinte du système nerveux central et beaucoup plus fréquente que l’atteinte du système nerveux périphérique.
2.Devant des nécroses digitales, il faut également exclure des emboles fibrino-cruriques qui sont généralement à point de départ intracardiaque. Cependant chez ce patient, il n’y a pas de thrombus intracardiaque mis en évidence.
3.Le diagnostic d’une hémopathie favorisant les thromboses est à évoquer devant une perturbation hématologique à la biologie. La numération-formule sanguine chez ce patient n’est pas en faveur de ce diagnostic.
4.La présence d’une cryoglobulinémie est le premier diagnostic à évoquer, car ce patient présente une hépatite C (VHC) avec une atteinte à la fois cutanée, neurologique et rénale.
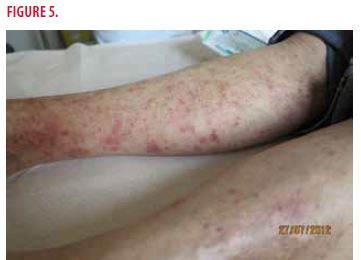
Une recherche de cryoglobulines met en effet en évidence une cryoglobulinémie de type II avec un fort composant monoclonal IgM à 5g/L et une activité anti-IgG polyclonale souvent associée au VHC. De plus, le patient présente un purpura chronique accompagné de dermite ocre souvent présent dans les cryoglobulinémies associées au VHC (Figure 5).
La ponction-biopsie rénale montre une glomérulonéphrite membranoproliférative avec des éléments inflammatoires obstruant les capillaires glomérulaires associés à des thrombus hyalins (étant des dépôts d’IgM monoclonale). Le patient est dès lors traité par trois échanges plasmatiques, des bolus de corticoïdes et de rituximab ainsi qu’une héparinothérapie. Le traitement améliore les lésions cutanées et rénales. Malheureusement, le patient devra subir une greffe hépatique suite à une évolution cirrhogène de l’hépatite C un an après.
Qu’est-ce qu’une cryoglobulinémie ?
Les cryoglobulines sont des immunoglobulines sériques qui précipitent au froid et qui solubilisent lors du réchauffement. On distingue trois types de cryoglobulinémies.
Il est intéressant à retenir que plus le composant monoclonal est important plus on oriente la clinique vers un aspect de thromboses plutôt que d’une vascularite. Dans les cryoglobulinémies de type I avec une IgM monoclonale unique, on aura essentiellement des thromboses associées à une lymphoprolifération B. Les cryoglobulinémies mixtes de type II avec un composant polyclonal et un composant monoclonal se retrouvent surtout dans les lymphoproliférations B et dans les maladies auto-immunes, néoplasiques et l’hépatite C. Les causes des cryoglobulinémies de type III, qui sont complètement polyclonales, sont comparables à celles du type II. Toutefois, 15% des cryoglobulinémies ne présentent aucune cause clairement élucidée et sont dites « essentielles ».
La biopsie cutanée est fondamentale pour différencier la vascularite, que l’on retrouve essentiellement dans les types II (qui ne sont pas à fort composant monoclonal) et III, de la thrombose que l’on retrouve surtout dans les types I et II (avec fort composant monoclonal). À l’analyse histologique des lésions de purpura palpable des cryoglubulinémies de type II (essentiellement mixtes) et des cryoglobulinémies de type III, on retrouve souvent une vasculite leucotyoclasique et des dépôts vasculaires d’IgG, IgM et de C3 en immunofluorescence directe. À la biologie sanguine, la consommation du complément est constante avec diminution du C1q, C2, C4 et CH50. Par contre, le taux de C3 est paradoxalement souvent normal. Le traitement en urgence est le traitement de l’ischémie avec les vasodilatateurs, tels que les inhibiteurs calciques et les analogues de la prostacycline (Iloprost®, Ventavis®), ainsi que les antiagrégants et/ou anticoagulants. Par ailleurs, le taux d’immunoglobulines doit être diminué par des échanges plasmatiques et des traitements immunosuppresseurs tels que les corticoïdes systémiques ou le rituximab. Il faudra traiter la cause dans un second temps.
Quel autre diagnostic différentiel ne faut-il pas oublier devant des nécroses cutanées ?
Il faut aussi penser aux cryofibrinogénémies. Cette cause de vasculopathie thrombosante reste pourtant mal connue.
Les cryofibrinogénémies sont secondaires à la précipitation au froid de protéines plasmatiques (fibrinogène, fibrine, fibronectine, facteur VIII, …), à la différence des cryoglobulinémies qui sont des immunoglobulines qui précipitent dans le sérum.
Au niveau de la physiopathologie, il y a des anomalies de la fibrinolyse avec augmentation des inhibiteurs de la fibrinolyse et un temps de lyse des euglobulines allongé engendrant une vasculopathie des vaisseaux de petits et moyens calibres.
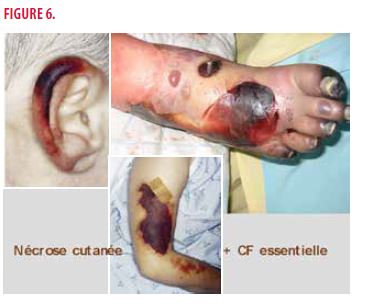
Cliniquement, la peau est l’organe cible avec des manifestations inaugurales dans > 80% des cas. On retrouve le plus souvent des nécroses cutanées (Figure 6) et des ulcérations (36-56%) avec une sensibilité extrême au froid chez 53-88% des patients. Il peut y avoir aussi des atteintes systémiques diverses telles que des arthralgies, myalgies, neuropathie périphérique, atteinte rénale, thrombose artérielle ou veineuse.
Les cryofibrinogénémies sont soit d’origine primitive soit secondaire. Dans les causes de cryofibrinogénémies secondaires, 1/3 sont des lymphomes/cancers, 1/3 des infections et 1/3 des connectivites. On note souvent une hyper α2-globulinémie à l’électrophorèse des protéines.
Parmi les éléments diagnostiques, on retient :
- une présentation clinique suggestive;
- la présence d’un cryofibrinogène plasmatique;
- une biopsie cutanée évocatrice : vasculopathie pauci-inflammatoire avec occlusion des vaisseaux par des thrombi éosinophiles et des dépôts de cryofibrinogène
- à l’imagerie, on retrouve une vasculopathie thrombosante des artères de moyens calibres;
- absence de cryoglobulinémie;
Le traitement des formes sévères reprend les fibrinolytiques intraveineux tel que l’altéplase (10mg/24h) ou la streptokinase (25000 U/24h) avec un relais par stanozolol (4 à 8mg/j per os) en traitement préventif des récidives. Les corticoïdes (0.5 à 1mg/kg/j) et immunosuppresseurs peuvent être prescrits mais n’ont pas fait preuve d’efficacité. L’aspirine à dose anti-agrégante et une héparinothérapie doivent être prescrites en cas de thrombose veineuse associée. Les récidives sont très fréquentes dans 42-76% des cas, dont la nécessité de se protéger du froid.
Caractéristiques cliniques de deux maladies de système rares avec manifestations cutanées
Cas clinique n° 3
Il s’agit d’une patiente de 58 ans en surcharge pondérale avec hypertension artérielle depuis 3 ans et un syndrome du canal carpien droit. La patiente a eu 3 grossesses sans particularité et a fait 3 fausses couches précoces. Une première hospitalisation a lieu en rhumatologie pour acrocyanose et polyalgies. La patiente présente alors des douleurs lombaires et du bassin d’horaire inflammatoire, une hypertrichose, un hippocratisme digital, une hépatosplénomégalie modérée et des adénopathies axillaires bilatérales.
Des prélèvements sanguins sont effectués. On retrouve :
- un pic monoclonal IgG lambda à 3.5 g/l
- des anticorps anticardiolipines positifs
- des anticorps anti-noyaux (AAN) à 1/160
- une cryoglobulinémie positive
- absence d’ANCA
- une calcémie normale
- ß2-microglubulinémie et LDH élevés
- TSH élevée
- Créatininémie normale
Parmi les examens complémentaires, une radiographie du squelette axial montre des lésions multiples ostéocondensantes du rachis lombaire, du bassin et des genoux. Le myélogramme est normal et la scintigraphie osseuse présente une absence de fixation.
Quels sont les diagnostics à évoquer devant ce tableau clinique ?
1. Un lupus systémique est peu probable. En effet, même si la patiente a des anticorps antinucléaires mais à taux faibles et une cryoglobulinémie, on ne peut expliquer les lésions ostéocondensantes par ce diagnostic.
2. Un lymphome, notamment un lymphome extra-nodal, est à évoquer.
3. Les lésions osseuses sont ostéolytiques dans la sarcoïdose. Vu les lésions osseuses ostéocondensantes, ce diagnostic est à exclure.
4. Il faut penser à exclure un POEMS syndrome. Néanmoins, la polyneuropathie est systématique dans un POEMS. On ne peut donc pas faire un diagnostic de POEMS sans sa présence.
Toutefois, un an plus tard, la patiente revient dans le service de dermatologie pour une altération de l’état général (- 20kgs en 1 an) et des paresthésies bilatérales des membres inférieurs.
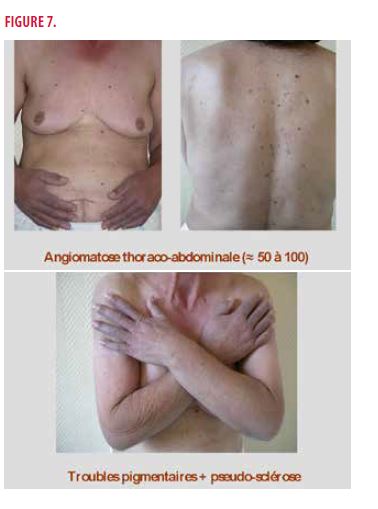
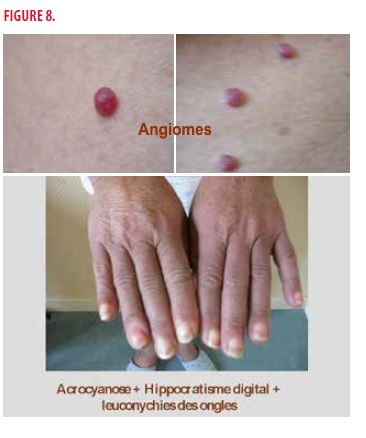
À l’examen clinique général, on retrouve des hypoesthésies en chaussettes avec abolition des réflexes achilléens, des adénopathies axillaires bilatérales associées à une hépatosplénomégalie, une tension artérielle à 130/60mmHg et une fréquence cardiaque à 90 battements/min. Au niveau cutané, on observe une angiomatose thoraco-abdominale (avec 50-100 angiomes tubéreux), une acrocyanose avec un hippocratisme digital et des leuconychies des ongles, des zones de pseudo-sclérose cutanée et des zones d’hyperpigmentation avec une hypertrichose en regard (Figures 7 et 8).
L’électromyogramme montre la présence d’une neuropathie démyélinisante bilatérale et symétrique sans atteinte axonale. La capillaroscopie est normale. Une radiographie du rachis montre une ostéocondensation de T7-T8 correspondant à des plasmocytomes multiples (Figure 9) et la scintigraphie osseuse présente une hyperfixation du sternum et L1. Finalement, le taux de VEGF sanguin est très élevé à 1949 pg/ml (normal = 273).
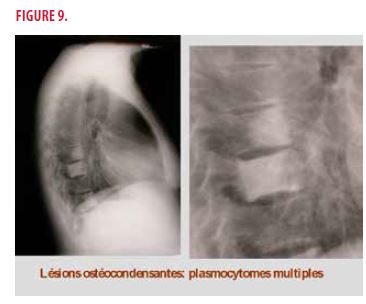
Devant ce tableau clinique, le diagnostic retenu est le diagnostic de POEMS syndrome. La patiente est dès lors traitée par une autogreffe de cellules souches périphériques, conditionnée par melphalan à haute dose. Chez cette patiente, ce traitement entraînera l’amélioration de l’état général, un gain d’autonomie, une diminution des douleurs osseuses, de l’acrocyanose et du nombre d’angiomes mais pas d’amélioration de la neuropathie.
Qu’est-ce que le POEMS syndrome ?
Ce syndrome a été décrit par Crox au Japon en 1965. L’épidémiologie reste totalement inconnue. Par contre, on retrouve un sex ratio de 2 hommes/1 femme et une survie médiane de 8 ans.
POEMS est l’acronyme anglais de
- Polyneuropathy : polyneuropathie périphérique
- Organomegaly : organomégalie
- Endocrinopathy : endocrinopathie
- Monoclonal gammapathy : dysglobulinémie monoclonale
- Skin changes : anomalie cutanée
Dans le POEMS syndrome, la polyneuropathie est constante et inaugurale dans 50% des cas. Elle est périphérique, symétrique et localisée en distal des membres inférieurs. Elle est d’abord sensitive puis motrice, démyélinisante puis axonale et s’accompagne d’une hyperprotéinorachie sans hypercytose à l’analyse du liquide céphalo-rachidien. Il peut également y avoir un œdème papillaire.
La gammapathie monoclonale peut être de type IgG ou IgA et est presque toujours de sous-type lambda. Elle est associée à une dyscrasie plasmocytaire à l’origine du syndrome. Cette dyscrasie est associée soit à un myélome, soit un plasmocytome, soit une MGUS (gammapathie monoclonale bénigne) ou encore une maladie de Castelman. Les lésions osseuses, qui en découlent, sont essentiellement ostéocondensantes, très rarement ostéolytiques.
Concernant les anomalies cutanées, on retrouve :
- des angiomes tubéreux (86%)
- une hyperpigmentation (76%)
- un épaississement cutané et une sclérose (57%)
- une acrocyanose (57%) et des ongles blancs (38%)
- un hippocratisme digital (24%)
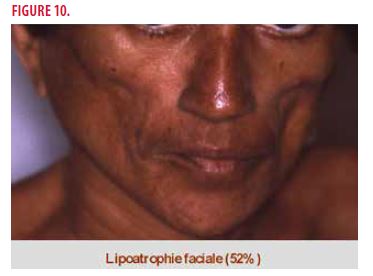
- une lipoatrophie faciale (52%)
- un livédo infiltré et nécrotique (28%)
La lipoatrophie faciale du POEMS syndrome est fréquente mais reste totalement inexpliquée (Figure 10).
Au niveau thérapeutique, thalidomide, lénalidomide et l’association dexaméthasone-melphalan ont également démontré une efficacité.
Cas clinique n°4
Il s’agit d’un homme de 70 avec comme antécédents personnels, une pneumopathie à germes atypiques avec pachypleurite en 2007, une phako-exérèse de l’œil droit suivie d’une uvéite en 2008, une arthrite des poignets avec carpite peu destructrice (facteur rhumatoïde et anticorps anti-citrullines négatifs) traitée par méthotrexate puis léflunomide en 2009, un épaississement de l’aorte abdominale étiquetée d’athérosclérose et un adénocarcinome de la prostate traité par Ablatherm en 2012. Il n’a pas d’allergies connues, ne boit pas, ne fume pas et ne voyage pas.
L’histoire de l’affection actuelle débute en 2012 avec un œdème de la cheville gauche associé à un érythème et une sclérose cutanée qui s’étend au niveau de la cheville droite 6 mois plus tard. Le patient signale une perte de 4kgs en 6 mois et la prise de nombreuses cures d’antibiotiques pour des diagnostics d’érysipèles récidivants.
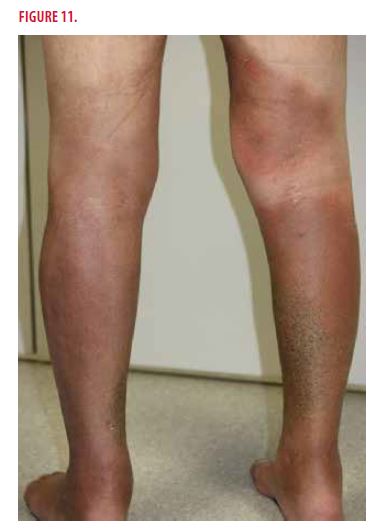
À l’examen clinique, on observe une infiltration cutanée profonde avec de vastes placards inflammatoires des membres inférieurs (Figure 11). Le patient est par ailleurs en bon état général. On ne palpe pas d’organomégalie, ni d’adénopathies périphériques.
À l’examen biologique, on retrouve, une numération-formule sanguine normale, un discret pic monoclonal d’IgG kappa, des dosages normaux des sous-classes d’Immunoglobulines, une CRP à 39 mg/l, une fonction rénale et un bilan hépatique normaux. Le bilan immunologique est négatif.
Quelles sont les hypothèses diagnostiques à évoquer devant ce tableau clinique ?
1.Morphées
2.Borreliose tertiaire
3.Lymphome cutané
4.Autre
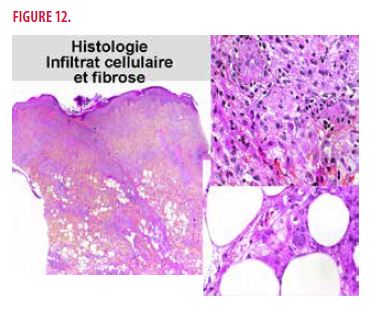
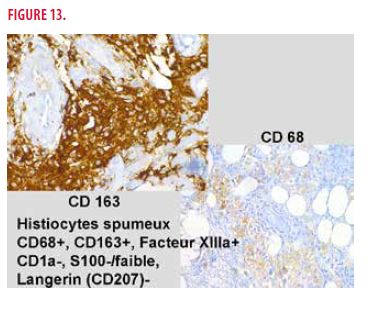
Devant ce tableau très peu spécifique, une biopsie cutanée est réalisée. On retrouve un infiltrat cellulaire très importante associé à de la fibrose. À plus fort grossissement, on observe des histiocytes spumeux et des cellules géantes plurinucléées (Figure 12). L’immunomarquage montre que les histiocytes spumeux sont CD68 +, CD 163 +, Facteur XIIIa +, CD1a -, Langerin (CD207) - et S100 – (Figure 13).
Les examens complémentaires suivants sont réalisés :
Les épreuves fonctionnelles respiratoires (EFR) montrent des volumes diminués. L’échographie cardiaque est normale et l’IRM cérébrale ne présente pas d’anomalie. Au PET-CT, on voit des fixations multiples notamment osseuses sur les humérus, les fémurs, les tibias, les poignets, le sternum et la clavicule droite, une fixation rétro-orbitaire bilatérale, ainsi qu’une fixation de l’aorte, des surrénales et une fixation cutanée des 2 membres inférieurs.
Le diagnostic final retenu, devant l’infiltrat histiocytaire à la biopsie cutanée, l’atteinte osseuse, rétro-orbitaire et péri-aortique et les antécédents de pachypleurite et de polyarthrite, est la Maladie d’Erdheim-Chester. Ce patient reçoit dès lors un traitement par Interferon pégylé (135 µg/semaine).
Qu’est-ce que la maladie d’Erdheim-Chester ?
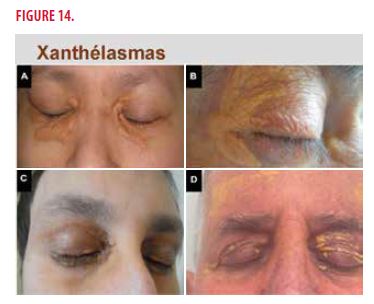
Au niveau cutané, l’atteinte la plus fréquente est le xanthélasma (25%). La plupart du temps, le xanthélasma est une lésion bénigne qui peut refléter le signe d’une hypercholestérolémie-hypertriglycéridémie sous-jacente.
Un xanthélasma, comme point d’appel d’une maladie d’Erdheim-Chester, est tout à fait exceptionnel mais ne doit pas être méconnu du clinicien. Dans cette affection systémique, le xanthélasma est caractérisé par des dépôts jaunâtres des paupières bilatéraux, circonférentiels et symétriques (Figure 14). Chez un malade avec xanthélasma, chez qui on suspecte une maladie d’Erdheim-Chester, il faut biopsier le xanthélasma afin de rechercher la mutation BRAF V600E.
Chez ce patient, l’analyse faite au niveau des biopsies cutanées montre une maladie d’Erdheim-Chester BRAF V600E sauvage (50% sont BRAF mutées). L’analyse des autres voies MAPK-kinases (NRAS, Kirsten RAS, MAP2K1) sont de nos jours également réalisées systématiquement et des mutations sont présentes dans 80% des maladies d’Erdheim-Chester.
Les autres lésions cutanées observées sont des plaques infiltrées des jambes, du dos et du tronc, des lésions papulo-nodulaires et une érythrodermie. Cette maladie peut provoquer également de multiples atteintes d’organe (Figure 15) et est souvent associée à une histiocytose langerhansienne. Actuellement, le traitement de premier choix est constitué par les thérapies ciblées : anti-MEK et anti-BRAF.

Trois nouvelles entités dans les maladies de système
Cas clinique n° 5
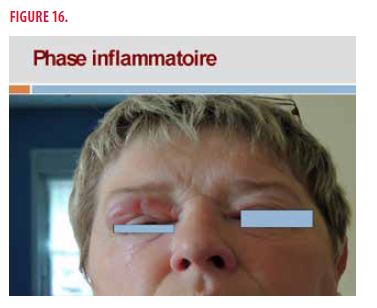
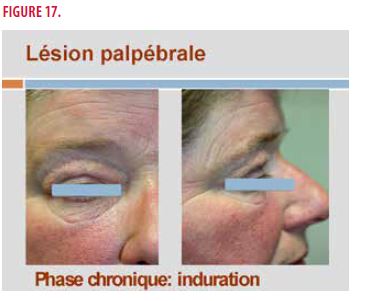
Il s’agit d’une enseignante de 55 ans. Dans ses antécédents personnels, on note un diabète de type II, une vasculite urticarienne présente 8 ans auparavant associée à un syndrome de Gougerot-Sjögren (avec syndrome sec et anticorps anti-SSA positifs) traitée par hydroxychloroquine 400mg/j. Actuellement, elle présente depuis 2 ans un œdème intermittent inflammatoire des paupières, cortico-sensible, se transformant progressivement en induration permanente. À l’examen clinique, on retrouve un œdème inflammatoire lors de la phase aiguë (Figure 16) et une induration très ferme en dehors des poussées aiguës (Figure 17).
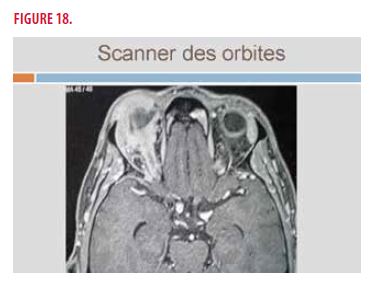
Au scanner orbitaire, on note une infiltration importante des tissus périorbitaires (Figure 18).
Le bilan auto-immun montre :
- AAN faibles : 1/320 de type moucheté
- anticorps anti-SSA négatifs
- dosage C1 inhibiteur, C3,C4,CH50 normaux
- fonction thyroïdienne (TSH,T3,T4) normale et absence d’anticorps antithyroïdiens
Quels sont les diagnostics à évoquer devant un tel tableau clinique ?
1.L’angio-œdème bradykinique héréditaire de type III est caractérisé par des épisodes de douleurs abdominales et des œdèmes de la face et des extrémités transitoires et récidivants. Il prédomine chez la femme et est favorisé par les pilules œstro-progestatives et par les grossesses. Toutefois ce diagnostic est à exclure chez cette patiente, car les œdèmes bradykiniques ne sont pas uniquement palpébraux et ne s’accompagnent pas d’une induration permanente.
2.La maladie de Morbihan est caractérisée par un œdème érythémateux chronique non induré préférentiellement du front et des paupières. Dans le cas de la patiente, un œdème de rosacée paraît peu probable vu l’induration permanente des lésions palpébrales.
3.Un lymphome palpébral est à évoquer surtout lorsqu’il y a un syndrome de Gougerot-Sjögren associé. De plus, un lymphome peut présenter des poussées inflammatoires et une infiltration.
4.Une sarcoïdose n’est pas exclue vu les lésions indurées fixes. Par contre, les poussées inflammatoires et l’absence d’aspect lupoïde ne plaident pas pour ce diagnostic.
5.La maladie associée aux IgG4 est à prendre en compte dans le diagnostic différentiel. En effet, les localisations palpébrale et orbitaire sont très fréquentes dans ce syndrome.
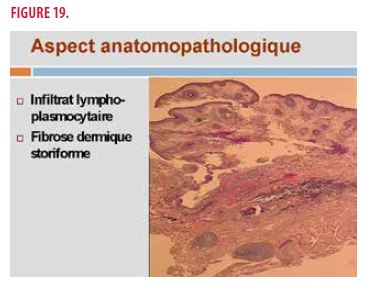
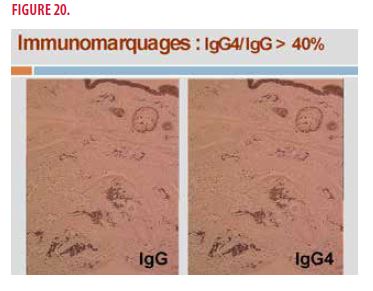
Une biopsie cutanée s’impose. Le diagnostic final retenu est une maladie associée aux IgG4. En effet, on retrouve un infiltrat lymphoplasmocytaire (Figure 19) et une fibrose dermique storiforme associé à un rapport IgG4/IgG tissulaire > 40% (Figure 20).
Pourquoi est-ce une maladie associée aux IgG4 ?
La topographie est compatible avec à l’analyse histologique une fibrose storiforme et un infiltrat lymphoplasmocytaire +- éosinophiles et un rapport tissulaire IgG4/IgG > 40%. On retrouve également une augmentation des IgG4 sériques > 135 mg/dl. Cette augmentation est par contre inconstante et non spécifique (elle peut aussi apparaître au cours d’infections ou de maladies auto-immunes).
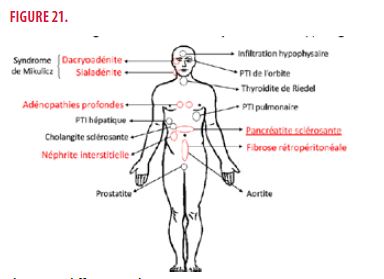
En résumé, la maladie associée aux IgG4 est une entité récemment décrite caractérisée par la présence d’une ou plusieurs atteintes fibro-inflammatoires d’organe associées le plus souvent à une élévation des IgG4 sériques. Il existe différents phénotypes (Figure 21: associations préférentielles) au sein de la maladie associée aux IgG4 repris ci-dessous :
- PHB : pancréato-hépato-biliaire
- RPA : rétropéritonéal et aortique
- CCU : cervico-céphalique uniquement
- MS : syndrome de Mikulicz (atteinte des glandes salivaires et lacrymales) + atteinte systémique
La maladie associée aux IgG4 est très fortement cortico-sensible. Le méthotrexate, la ciclosporine et l’azathioprine peuvent également être administrés. Le rituximab a montré une bonne efficacité (>90%) mais on observe des rechutes dans 42% des cas. En outre, le thalidomide a été proposé dans l’atteinte cutanée isolée.
Cas clinique n°6
Il s’agit d’une femme de 22 ans d’origine marocaine, née de parents consanguins. Dans ses antécédents personnels, on retrouve une surdité bilatérale acquise apparue à l’âge de 4 ans, appareillée à l’âge de 10 ans et bénéficiant de la pose d’un implant cochléaire à l’âge de 25 ans. Les antécédents familiaux sont sans particularité et elle ne prend pas de traitement. Il n’y a pas de prise de toxiques, aucune allergie connue et pas de voyage récent.
Elle consulte pour des nappes hyperpigmentées des faces internes des cuisses apparues à l’âge de 17 ans, asymptomatiques mais inesthétiques et en extension progressive (Figure 22).
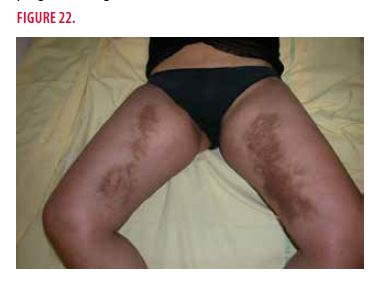
À l’examen clinique, on observe une Infiltration profonde surmontée de vastes placards hyperpigmentés des faces internes des cuisses pour lesquels différents traitements locaux ont été testés auparavant mais sont restés inefficaces (dermocorticoïdes, kératolytiques, hydroquinone, …). La jeune femme est en bon état général (Poids= 58 kg, Taille= 1,58 m, BMI=23), absence d’organomégalie et absence d’adénopathies péripheriques. Par contre, on palpe un blindage des régions inguinales englobant les aires ganglionnaires (Figure 23). Elle présente également un gérontoxon bilatéral des yeux.

Une biopsie cutanée est réalisée et retrouve un épiderme avec une hyperkératose, une acanthose et une augmentation des dépôts de mélanine dans les kératinocytes basaux associée à une fibrose étendue du derme et de l’hypoderme avec un infiltrat mononucléé interstitiel composé d’histiocytes CD68+, S100+, CD1a- (Rosai-Dorfman), empéripolèse et de nombreux dendrocytes CD34+ et facteur XIIIa+.
À la biologie, on note une carence martiale sans saignement occulte, un syndrome inflammatoire modéré (VS = 88 mm, CRP = 106 XX), une hypergammaglobulinémie polyclonale (= 19 g/L), des anticorps anti-nucléaires positifs (AAN = 1/160), des anticoprs anti-ADN négatifs, les sérologies borréliose négatives, une sérologie leishmaniose et syphilis négatives.
Une échographie et une IRM des tissus mous des faces internes des cuisses montrent un épaississement et une infiltration hyperéchogène du tissu sous-cutané de la région pubienne, inguinale et des cuisses, associés à un œdème. On constate une absence d’adénopathies d’allure pathologique, absence d’anomalies des fascias et de signes de myosite.
Quelles sont les hypothèses diagnostiques retenues devant ce tableau clinique ?
1.Morphées
2.Borréliose tertiaire
3.Lymphome cutané
4.Autre
Une prise en charge initiale avec un traitement d’épreuve par amoxicilline et ceftriaxone est mis en place sans grand succès. On assiste d’ailleurs à l’extension de l’infiltration en profondeur jusqu’aux espaces intermusculaires des cuisses. Le diagnostic alors retenu est celui de morphées profondes et un traitement par corticoïdes et immunosuppresseurs est proposé. Mais la patiente, enceinte, est perdue de vue…
Finalement, le diagnostic de syndrome H est suspecté devant l’origine ethnique de la patiente, l’aspect lésionnel, le caractère symétrique et évolutif des placards indurés hyperpigmentés apparus au cours de la 2e décennie, l’infiltration pelvienne et les signes associés (surdité, petite taille). Le diagnostic est confirmé par l’analyse génétique retrouvant la mutation P324L du gène SLC29A3.
Qu’est-ce que le syndrome H ?
Le syndrome H comprend les caractéristiques cliniques suivantes :
- Hyperpigmentation cutanée et Hypertrichose
- Hépatosplénomégalie
- Surdité (Hearing loss)
- Hypogonadisme
- Anomalies cardiaques (Heart anomalies)
- Petite taille (Low Height)
- Hyperglycémie
Il s’agit d’une génodermatose autosomique récessive avec mutation du gène SLC29A3 codant pour la protéine de transport nucléotidique hENT3 (>17 mutations ont été identifiées). Dans la population palestinienne d’origine arabe, on retrouve 1% de porteurs sains des deux plus fréquentes mutations (G437R,G427S). La maladie reste néanmoins ubiquitaire (Espagne, Japon, Chine, …). Le diagnostic est difficile, car il s’agit d’une maladie rare et peu connue avec une importante variabilité phénotypique. Il existe beaucoup de formes pauci-symptomatiques et l’accumulation des symptômes vient avec l’âge. L’évolution reste méconnue.
Au niveau dermatologique, on retrouve dans 68% des cas, des placards hyperpigmentés et indurés sclérodermiformes, symétriques et apparaissant au cours de la 1ère ou 2ème décennie et d’aggravation progressive (Figure 24). Ils ont parfois un aspect ichtyosique et sont parfois associés à une hypertrichose. La localisation préférentielle de ces placards se situe sur la moitié inférieure du corps au niveau des faces internes des cuisses, des jambes, de l’abdomen, de la région pelvienne et de la région lombaire avec un respect des genoux et fesses. On observe également des varices et des télangiectasies faciales.

Parmi les signes extra-cutanés retrouvés dans le syndrome H, on a :
-Petite taille 49%
-Rétraction en flexion des doigts et des orteils 56%
-Surdité neurosensorielle 53%
•Congénitale ou acquise
•Aggravation progressive
-Hépatosplénomégalie 43%
-Adénopathies inguinales 24%
-Anomalies endocriniennes
•Masses scrotales et labiales
•Micropénis
•Diabète
•Gynécomastie
-Malformations cardiaques
•Cardiomégalie
•Prolapsus mitral
•Anomalies du septum auriculaire ou ventriculaire
•Agénésie de la Veine cave inférieure
-Anomalies oculaires
•Exophtalmie
•Arc sénile (Gérontoxon)
•Dilatations des vaisseaux de la sclère
-Anomalies ostéo-articulaires (Figure 25)
•Camptodactylie
•Déformations articulaires
•Hallux valgus
•Ostéoporose
Tous ces signes sont le plus souvent acquis.
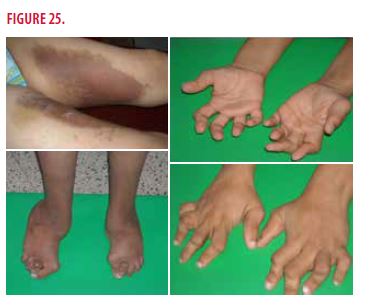
Au niveau des examens complémentaires, la biologie sanguine retrouve un syndrome inflammatoire (élévation de la VS (>100), élévation de la CRP, anémie microcytaire et des anomalies endocriniennes (déficit en GH, hypogonadisme hypergonadotrope, azoospermie). Au scanner abdomino-pelvien, on observe le plus souvent une infiltration et un épaississement sous-cutanés ainsi que des adénopathies inguinales.
Actuellement, il n’existe aucun traitement spécifique. La colchicine associée à la PUVAthérapie et la corticothérapie générale peuvent parfois donner des améliorations partielles. Le tocilizumab (anticorps anti-IL6) semble prometteur avec amélioration de l’infiltration cutanée et de la qualité de vie et amélioration du syndrome inflammatoire biologique et du diabète.
Cas clinique n°7
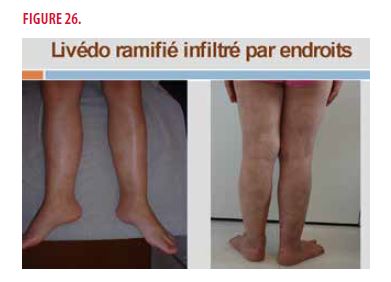
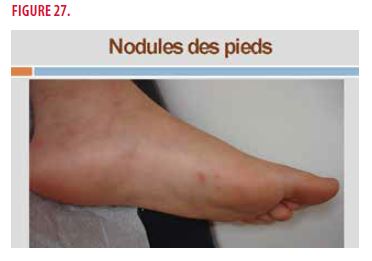
Il s’agit d’une jeune fille de 11 ans. Dans ses antécédents personnels, on note de multiples infections ORL à partir de 6 mois de vie, un livédo ramifié des membres inférieurs depuis l’âge de 4 ans, une occlusion de l’artère centrale de la rétine gauche avec une acuité visuelle de 2/10 à 10 ans, un accident ischémique du tronc cérébral avec paralysie de la 3ème paire crânienne gauche à 7 ans et une hypogammaglobulinémie à 5g/l. Les antécédents familiaux sont sans particularité. Elle suit un traitement par clopidogrel 1co/J en prophylaxie thrombo-embolique. À l’examen clinique, on observe : un livédo ramifié des membres inférieurs, infiltré par endroits, et des nodules au niveau des pieds et des chevilles (Figures 26 et 27).
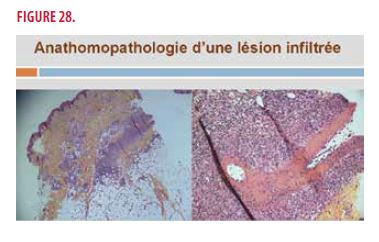
Une biopsie cutanée d’une des lésions infiltrée est réalisée et montre une image de péri-artérite noueuse cutanée permettant de poser ce diagnostic (Figure 28).
Qu’est-ce que la péri-artérite noueuse ?
Les facteurs déclenchants de la péri-artérite noueuse cutanée (PANc) peuvent être les infections (Parvovirus B19, hépatites B et C, CMV, les infections à Streptocoques β-hémolytique, la tuberculose, …), les maladies inflammatoires du tube digestif et les médicaments (minocycline). Cependant, dans la grande majorité des cas, on ne retrouve pas de facteur déclenchant.
Chez cette petite patiente, il existe une mutation récessive du gène CECR1, codant pour l’adénosine déaminase 2 (ADA2), détectée initialement dans des formes familiales associant PAN cutanée et PAN systémique. Cette mutation récessive du gène CECR1 provoque un déficit en ADA2. Le diagnostic final retenu est une maladie avec déficit en ADA2.
Cette maladie avec déficit en ADA2 débute dans l’enfance chez des enfants sans histoire familiale de vascularite et donne les caractéristiques cliniques suivantes :
- Lésions cutanées à type de livédo, ulcérations, nodules cutanés isolés mais avec fréquentes infections bactériennes et/ou virales
- Nécroses digitales
- Fièvre, myalgies, arthralgies
- AVC ischémiques ou hémorragiques
- Neuropathie périphérique
- Hépatosplénomégalie avec hypertension portale
- A la biologie, on a souvent un déficit en lymphocytes B avec des taux bas d’IgM et cytopénies
- Taux plasmatiques bas d’ADA2 (mutation homozygote ou double mutations hétérozygotes du gène CECR1)
Le traitement comprend les anti-TNF et la transplantation de cellules hématopoïétiques.
Le concept du « syndrome des abcès aseptiques »
Cas clinique n°8
En 2004, un homme de 38 ans d’origine syrienne présente une aphtose bucco-pharyngée unipolaire, des douleurs abdominales et une diarrhée non glairo-sanglante.
À la colonoscopie, on retrouve des ulcérations iléales avec à l’histologie une hyperplasie lymphoïde sans granulome associé. L’oesogastroduodénoscopie montre des ulcérations aphtoïdes oesophagiennes et duodénales avec présence d’Helicobacter pylori et une inflammation non spécifique à l’histologie.
Le diagnostic d’une probable maladie inflammatoire chronique intestinale (MICI) est retenu. Le patient est alors traité par corticothérapie systémique qui montre une bonne efficacité clinique mais le patient devient vite cortico-dépendant nécessitant l’introduction d’azathioprine associé à la colchicine.
En 2005, une angio-IRM est réalisée pour des douleurs lombaires. Celle-ci met en évidence des anévrysmes sacciformes de l’aorte thoraco-abdomino-pelvienne et de la carotide primitive gauche.
Quelles sont les diagnostics à évoquer devant ce tableau clinique initial ?
1.Il faut penser à la maladie de Behçet vu l’aphtose unipolaire, l’atteinte vasculaire à type de vascularite artérielle et veineuse et le fait que le patient est originaire d’une population à risque. L’atteinte digestive est également possible dans cette affection (formes frontières entre les MICI et l’atteinte digestive de la maladie de Behçet).
2.La maladie de Takayasu est caractérisée par une artérite des artères de gros calibre chez des patients de jeune âge. Ce diagnostic est à évoquer, car il s’agit d’un sujet jeune et la maladie peut s’associer à une MICI.
3.La polychondrite atrophiante est une connectivite dont la caractéristique principale est l’inflammation récidivante des cartilages de l’oreille, du nez, du larynx et de l’arbre trachéo-bronchique évoluant par poussées. Ce diagnostic est à exclure chez ce patient vu l’absence d’atteinte des cartilages, de troubles de l’audition, de voix rauque…
En octobre 2006, on assiste à une rechute clinique avec réapparition des douleurs abdominales.
Les examens complémentaires suivant sont réalisés :
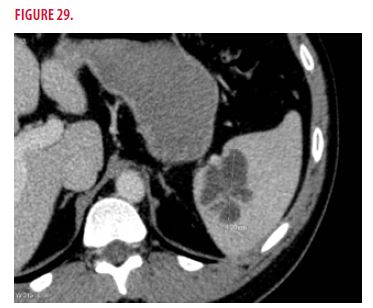
- le CT-scanner abdominal montre des lésions spléniques d’aspect kystique et polylobées compatibles avec un infarctus ou un abcès splénique (Figure 29).
- le PET CT-scan montre une co-localisation splénique isolée du traceur compatible avec un abcès ou une tumeur splénique.
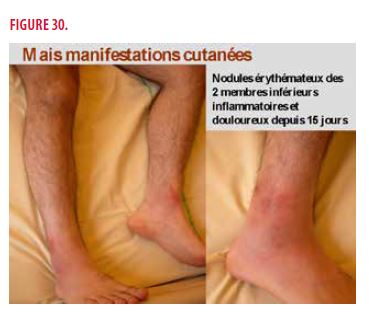
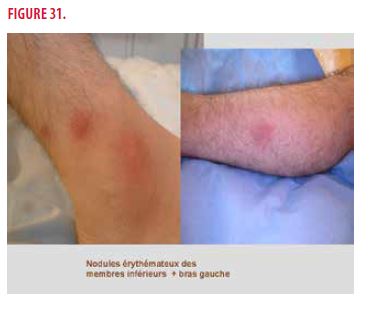
Finalement, une splénectomie diagnostique devait être programmée pour analyse anatomopathologique. Entre temps, des manifestations cutanées se sont présentées, à savoir des nodules érythémateux inflammatoires et douloureux des 2 membres inférieurs et du bras gauche plus accessibles à la biopsie cutanée (Figures 30 et 31). Un avis dermatologique est alors demandé.
Quels sont les diagnostics à évoquer à ce stade ?
1.Il faut penser à exclure des lésions infectieuses (mycobactéries, mycose profonde…) vu que le patient est sous traitement immunosuppresseur.
2.Il est possible qu’il s’agisse d’un Crohn métastatique mais les lésions sont normalement indolores.
3.La maladie de Behçet et de Takayasu peuvent donner des hypodermites et des panniculites se présentant sous la forme de nodules inflammatoires cutanés.
4.Un lymphome cutané est à exclure, car il y a certes une possibilité de lymphome splénique et cutané mais les anévrysmes sont difficiles à intégrer dans le tableau clinique.
5.Le diagnostic d’abcès aseptiques cutanés est à évoquer, car un abcès splénique n’est pas exclu et les nodules pourraient correspondre à la même lésion que celle retrouvée au niveau de la rate.
Une biopsie cutanée est réalisée. Elle montre un abcès aseptique à polynucléaires neutrophiles, de rares cellules géantes mais absence de granulome (Figures 32 et 33). Les colorations de PAS et Ziehl sont négatives.
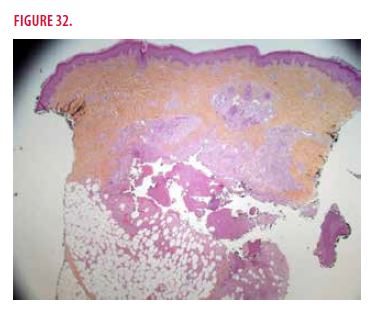
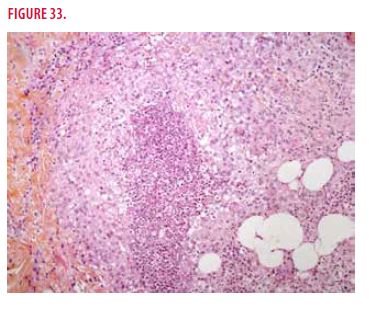
Devant la présence d’abcès aseptiques de la peau et de la rate, un « syndrome des abcès aseptiques » est confirmé. Une corticothérapie à forte dose (3 bolus de 1g puis 1mg/kg/j) est mise en route. À retenir qu’une antibiothérapie est inefficace. Une surveillance clinique de l’atteinte cutanée et de l’imagerie splénique est mise en place et permet de constater une régression rapide de l’atteinte cutanée en 20 jours sans récidive ainsi qu’une nette diminution en taille des images spléniques et disparition de la fixation au PET-scan.
En conclusion, ce patient présente un syndrome des abcès aseptiques sur une maladie de Crohn avec atteinte vasculaire ou maladie de Crohn et Takayasu.
Qu’est-ce qu’une maladie neutrophilique ou « Syndrome des abcès aseptiques » ?
Le syndrome des abcès aseptiques est caractérisé par de la fièvre (90%), des douleurs abdominales (67%) et une altération de l’état général (50%) apparaissant 5 mois en moyenne avant le diagnostic. Les organes atteints sont la rate (93%), les ganglions, le foie, les poumons, le pancréas et la peau (20%). Les rechutes sont fréquentes (60%).
Ces maladies systémiques sont notamment associées à des MICI, polyarthrite rhumatoïde, gammapathies monoclonales à IgA, syndromes myéloprolifératifs.
Un traitement immunosuppresseur en relais de la corticothérapie systémique est nécessaire chez 43% des patients. Le diagnostic sur l’atteinte cutanée permet d’éviter des gestes invasifs tel une splénectomie.
Affiliations
(1) Cliniques universitaires Saint-Luc, Dermatologie, avenue hippocrate 10, B-1200 Bruxelles, Belgique
(2) Hôpital Tenon, Dermatologie, Rue de la Chine 4, F-75020 Paris, France