Les nouveaux traitements de l'hémophilie
Cédric Hermans
Introduction
L’hémophilie représente la maladie hémorragique héréditaire la plus fréquente. Elle est la conséquence d’un déficit d’origine génétique complet (hémophilie sévère) ou partiel (hémophilie modérée ou mineure) du FVIII (FVIII) (Hémophilie A (HA)) ou FIX (FIX) (Hémophilie B (HB)) de la coagulation sanguine. Ce déficit entraîne des hémorragies qui surviennent surtout dans les articulations et les muscles (1).
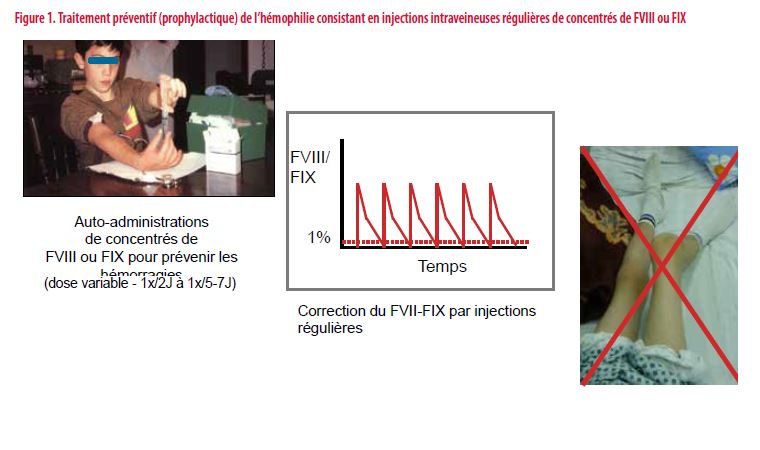
Le traitement des formes sévères repose sur le remplacement du FVIII ou FIX administré de façon régulière et préventive afin de prévenir les hémorragies et leurs conséquences locomotrices invalidantes (Figure 1). Ce traitement dit prophylactique est contraignant et partiellement efficace puisqu’il ne préserve pas totalement les articulations (2).
Le développement de concentrés modifiés de FVIII et FIX dotés d’une plus longue demi-vie, l’introduction d’un anticorps monoclonal bispécifique mimant l’action du FVIII ainsi que diverses stratégies modifiant l’équilibre de la coagulation représentent de nouvelles options de traitement (3). S’y ajoutent les premiers résultats prometteurs de la thérapie génique. Ces développements offrent aux patients hémophiles de nouvelles perspectives de traitement voire de guérison que cet article se propose de revoir (4).
Du drame du sang contaminé aux succès de la biotechnologie
Dans les années 50 et début des années 60, l’administration de sang total ou de plasma frais, comme sources de FVIII ou FIX, représentaient les seules options, contraignantes et peu efficaces, de traitement de l’hémophilie (5). En 1964, la découverte par Judith POOL que la décongélation lente du plasma permet d’obtenir une fraction enrichie en FVIII (cryoprécipité) a grandement facilité la prise en charge thérapeutique. Fin des années 60, les premiers concentrés de FVIII plasmatique de pureté croissante deviennent disponibles. Ils permettent d’envisager des traitements réguliers préventifs, même au domicile. En 1982, le premier cas de SIDA chez un patient hémophile est décrit. Le drame de la contamination des patients hémophiles par le HIV a motivé l’introduction de divers procédés de sécurisation des concentrés plasmatiques de FVIII et FIX. En 1989, le virus de l’hépatite C (VHC) est identifié, un virus par lequel un très grand nombre de patients exposés à des dérivés plasmatiques a été contaminé.
L’identification des gènes des FVIII et FIX a permis dans les années 90 le développement par biotechnologie des premiers concentrés synthétiques (recombinants) de FVIII et FIX. Des améliorations techniques ultérieures ont permis la mise à disposition de FVIII et FIX recombinants obtenus sans aucune addition de protéines humaines et animales éliminant en théorie tout risque infectieux.
Parallèlement, les concentrés issus du plasma ont été sécurisés par la multiplication de procédés chimiques ou physiques assurant la destruction ou l’élimination des agents infectieux, y compris les prions. Aucun cas de contamination des patients hémophiles par les virus tels que le HIV et le VHC n’a été rapporté au cours des deux dernières décennies.
Après de nombreuses années consacrées à garantir la sécurité infectieuse des concentrés de FVIII et FIX, les recherches récentes ont pour priorité et ambition de modifier les molécules de FVIII et FIX afin de faciliter le traitement de l’hémophilie, d’améliorer son efficacité et d’explorer d’autres options de traitements plus efficaces et moins contraignantes (6-9).
Traitements actuels de l’hémophilie : modalités et défis
Chez les patients hémophiles présentant une forme dite sévère (FVIII ou FIX : < 1 %), les hémorragies articulaires et musculaires peuvent survenir spontanément et sont plus fréquentes que chez les patients présentant une forme dite modérée (FVIII ou FIX : 1-5 %). Cette observation est à l’origine de l’introduction du traitement de substitution dit prophylactique, à savoir des injections régulières de facteurs dont le but est de maintenir une concentration de FVIII ou de FIX au-delà de 1 à 2 % et de réduire le risque d’hémorragies spontanées (10).
Le traitement de substitution de l’hémophilie par des concentrés de FVIII ou FIX, issus du plasma ou produits de façon synthétique par biotechnologie, se heurte à de nombreux défis :
1. Les concentrés de FVIII et FIX doivent être administrés par voie intraveineuse, ce qui se révèle souvent problématique chez les très jeunes enfants et constitue un obstacle à l’adhérence des patients
2. Les FVIII et dans une moindre mesure le FIX ont une demi-vie courte et sont rapidement éliminés de la circulation, ce qui impose des administrations fréquentes et alourdit la charge du traitement.
La pharmacocinétique et en particulier la vitesse d’élimination du FVIII et FIX, est très variable d’un patient à l’autre et se modifie au cours de la vie (11).
La correction de la concentration circulante du FVIII et du FIX obtenue par les injections régulières est la plupart du temps partielle, variable et fluctuante, rythmée par des pics de concentration dans le décours immédiat des injections et des vallées à distance des injections. Cette variabilité impose aux patients de nombreuses restrictions et complique le traitement.
Le FVIII est une molécule particulièrement immunogène dont l’administration peut entraîner le développement d’allo-anticorps neutralisants (inhibiteurs), la complication majeure et la plus redoutée du traitement de l’hémophilie.
Le traitement de substitution individualisé
De nombreuses études ont démontré l’importance d’individualiser et de personnaliser le traitement de substitution de l’hémophilie. Il s’agit d’adapter pour chaque patient le régime de traitement (dose de facteur, moment et fréquence des injections) en tenant compte de multiples variables (mode de vie, état articulaire, comorbidités et surtout le comportement pharmacocinétique du FVIII ou FIX injecté).
Aux études pharmacocinétiques classiques et fastidieuses (nécessitant de multiples prélèvements post-injection et en réalité rarement réalisées, surtout chez les enfants) se substitue actuellement l’approche bayésienne. Elle consiste, sur base d’un nombre très limité de dosages de FVIII et FIX issus de prélèvements réalisés à des moments variables sous traitement, d’extrapoler le comportement pharmacocinétique individuel à partir du profil obtenu au sein d’une population de patients. Il s’agit pour chaque patient de dériver par cette approche la demi-vie du FVIII ou FIX injecté, d’estimer l’exposition au traitement par calcul de l’aire sous-la-courbe et de définir le temps nécessaire pour atteindre certains seuils prédéfinis (10 %, 5 % et 1 %).
Plusieurs outils informatiques très performants ont été développés et validés. Certains tels My-PK Fit (12), sont spécifiques à un concentré, d’autres tels WAPPS-Hemo, peuvent être utilisés pour la plupart des concentrés de facteurs disponibles (13). Outre une approche plus rigoureuse du traitement de substitution qui tient compte des données pharmacocinétiques individuelles, ces outils sont utiles pour améliorer la compréhension par chaque patient des modalités et rationnel du traitement. Ils permettent aussi de promouvoir l’adhérence, réduire le nombre de saignements et encourager une meilleure utilisation des concentrés.
Les nouveaux traitements de l’hémophilie : substitutifs ou non-substitutifs
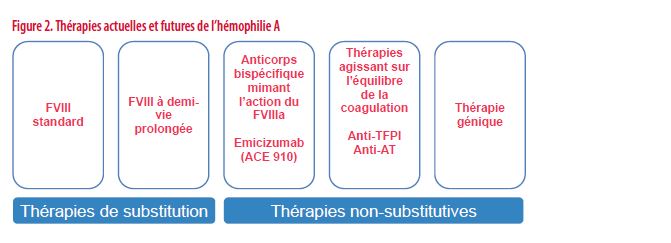
Au cours de la dernière décennie, le traitement de l’hémophilie a bénéficié de multiples innovations thérapeutiques dont certaines sont déjà largement implémentées. Attestant de cette évolution, on distingue actuellement les thérapies dites substitutives reposant sur l’administration de FVIII ou de FIX des thérapies dites non-substitutives (aussi qualifiées de disruptives) et indépendantes de l’administration de FVIII ou FIX (14).
Parmi les approches les plus avancées, il faut citer (Figures 2 et 3) :
1. Les concentrés de FVIII et FIX synthétiques modifiés et dotés d’une demi-vie prolongée.
2. L’immunothérapie avec le développement d’un anticorps monoclonal bispécifique mimant l’action du FVIII.
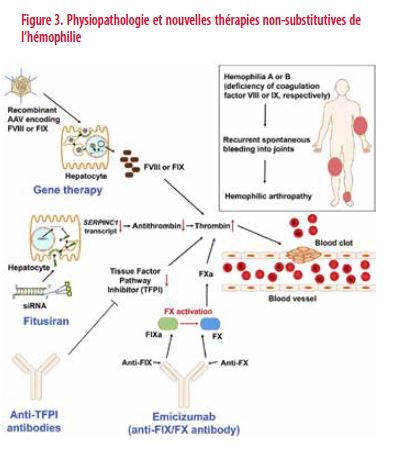
3. Les thérapies visant à modifier l’équilibre de la coagulation en agissant sur les inhibiteurs physiologiques de la coagulation. Il s’agit d’induire chez les patients hémophiles un état prothrombotique qui compense le déficit en facteur. Plusieurs approches sont en cours d’évaluation: la réduction de la production hépatique d’antithrombine (le principal inhibiteur de la coagulation) et l’inhibition ciblée du Tissue Factor Pathway Inhibitor (TFPI) (inhibiteur physiologique des facteurs VII et X).
4. La thérapie génique consiste à doter le foie de la propriété de synthétiser du FVIII ou du FIX.
Facteurs VIII et IX à demi-vie prolongée
Plusieurs stratégies sont utilisées pour allonger la demi-vie du FVIII et du FIX. La fusion avec l’albumine ou le fragment Fc des immunoglobulines permet au FVIII (Fc-FVIII) et FIX (Fc-FIX/Alb-FIX) de faire l’objet d’un recyclage intracellulaire (internalisation cellulaire via le récepteur Fc-RN et relargage ultérieur dans la circulation). C’est ce mécanisme qui explique la longue demi-vie tant de l’albumine que des immunoglobulines et dont bénéficient les FVIII et FIX après fusion. Une autre stratégie appliquée aux FVIII et FIX est la pégylation qui interfère avec l’élimination des facteurs et allonge leur demi-vie (Figure 4).
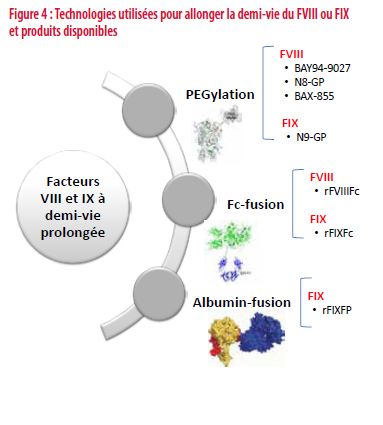
Par ces diverses techniques, il est possible d’allonger la demi-vie du FVIII de plus ou moins 40 %. Pratiquement, pour un patient qui aurait une demi-vie du FVIII de 12 heures avec un FVIII classique, on peut espérer un allongement à 19 heures avec un FVIII modifié. Cela s’explique par la fixation physiologique du FVIII au facteur von-willebrand dont la propre demi-vie limite l’allongement du FVIII modifié.
Même si cet allongement semble modéré, son impact sur la prise en charge des patients est loin d’être négligeable. Certains patients traités par 3 injections intraveineuses par semaine peuvent être traités 2 fois par semaine. Pour certains patients bien identifiés, il est même possible d’espacer davantage les injections (1x/5 jours voire davantage). Pour d’autres patients, en conservant le même rythme d’administration (3x/semaine), il est possible de maintenir une concentration basale de FVIII plus élevée et procurer aux patients une meilleure protection vis-à-vis des hémorragies.
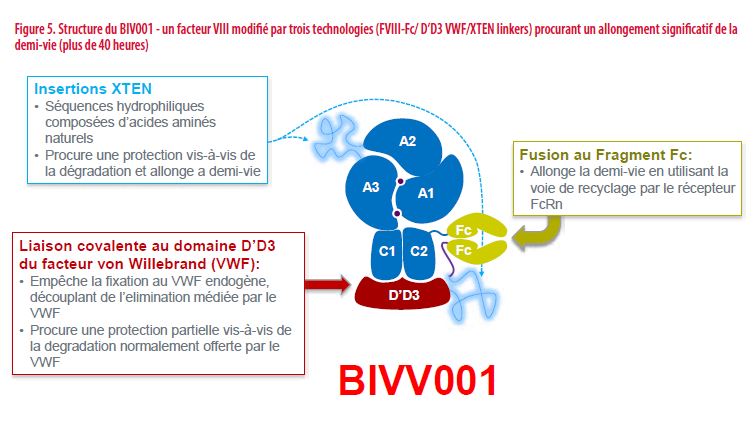
Pour davantage allonger la demi-vie du FVIII (au-delà de 19 heures), des modifications supplémentaires sont en cours de développement. Un Fc-FVIII auquel un fragment du facteur von willebrand (Domaine D’-D3) et des séquences peptidiques neutres (X-Ten) ont été fixées (FC-FVIII-VWF-XTEN) semble présenter un comportement pharmacocinétique bien plus favorable. Sa demi-vie de 37,6 à 44 heures devrait permettra des administrations hebdomadaires, une hypothèse actuellement évaluée dans des études cliniques (Figure 5).
Pour le FIX qui n’est pas lié à une protéine transporteuse comme le FVIII, sa modification par fusion (albumine ou fragment Fc) ou Pégylation, procure un allongement de la demi-vie bien plus important (4 à 5 fois) que pour le FVIII. Ceci permet de traiter les patients par des injections bien espacées, à des fréquences variables (1x/7-10-14 jours voire plus espacées) en fonction du type de FIX modifié et de la réponse de chaque patient.
L’immunothérapie de l’hémophilie par un anticorps monoclonal bi-spécifique
Cette approche originale fait appel à un anticorps monoclonal bispécifique appelé ACE910 ou Emicizumab (Hemlibra®) qui mime l’action du FVIII. Cet anticorps dispose de 2 sites de reconnaissance : le premier lui permet de se fixer au FIXa, le second au FX (Figure 6). Ce faisant, cet anticorps « mime » et assume l’action du FVIII au sein de la coagulation sanguine (15;16).
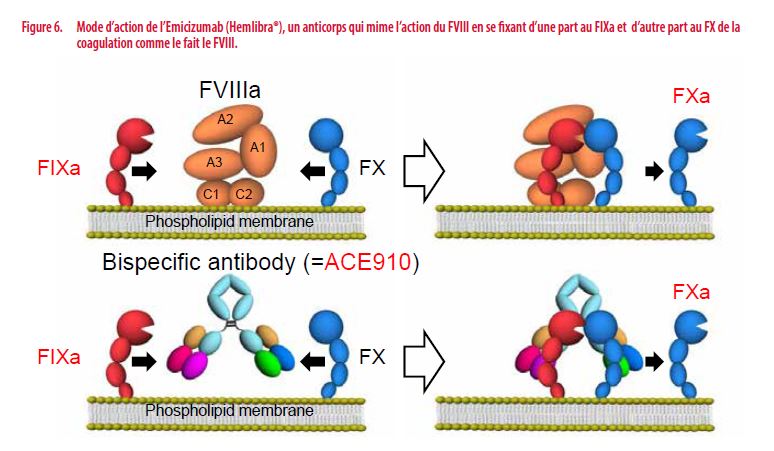
Compte tenu du fait qu’il ne s’agit pas de FVIII, cet anticorps n’est pas reconnu par les anticorps anti-FVIII et peut-être administré à des patients avec inhibiteur du FVIII. Cet anticorps n’entraîne également pas la formation d’anticorps anti-FVIII. Les autres avantages majeurs sont le fait que cette molécule peut être administrée par voie sous-cutanée et que sa demi-vie est extrêmement longue permettant des administrations peu fréquentes (une fois/semaine voire une fois/4 semaines) et le maintien d’une activité hémostatique constante après une phase d’imprégnation de 4 semaines.
De vastes études récentes ont démontré la très grande efficacité de ce traitement chez les patients hémophiles A adultes et enfants, avec et sans inhibiteur (17). L’effet hémostatique procuré par cet anticorps corrige partiellement mais pas totalement le déficit de coagulation des patients avec hémophilie A. Ces derniers sont donc susceptibles de présenter des accidents hémorragiques justifiant l’apport en FVIII intraveineux. De même, certains gestes invasifs ou interventions chirurgicales justifient également une supplémentation en FVIII pour garantir une hémostase suffisante (18).
En termes de sécurité, plusieurs patients avec inhibiteur traités conjointement par cet anticorps bispécifique et un agent court-circuitant le FVIII (FEIBA® - un concentré plasmatique des FIIa, FVIIa, FIXa et FXa) ont développé des thromboses dont une fatale (19). Il est totalement contre-indiqué de traiter simultanément un patient avec inhibiteur par Emicizumab et Feiba® mais de privilégier un autre agent de bypass du FVIII, le FVIIa (Novo Seven®). Quelques rares cas d’ADA (Anti-Drug Antibodies) ont été décrits.
Les agents « rebalançant » la coagulation sanguine
La stratégie originale consiste à réduire l’inhibition physiologique de la coagulation sanguine. Celle-ci est soumise à des mécanismes de contrôle assurés par des inhibiteurs physiologiques de la coagulation, à savoir l’antithrombine (AT) qui inhibe les FXa et FIIa (thrombine) et le TFPI (Tissue Factor Pathway Inhibitor) qui inhibe le FVIIa et FXa (20;21).
Le Fitusiran réduit, via une séquence d’ARN silencieux à tropisme hépatique interférant avec l’ARN endogène, la synthèse hépatique de l’AT dont la concentration circulante diminue de façon significative. La production majorée de thrombine qui en résulte permet de restaurer l’hémostase et d’empêcher les hémorragies chez les patients hémophiles. L’administration de Fitusiran par voie sous-cutanée entraîne une réduction de l’AT circulante de 80 % et une réduction significative des hémorragies (20).
Divers agents (aptamère, anticorps monoclonaux) inhibant le TFPI sont en cours de validation. Cette approche permet de majorer la production de thrombine et réduit les manifestations hémorragiques de façon significative (21;22).
Ces deux approches présentent plusieurs caractéristiques favorables : administration sous-cutanée, fréquence d’administration peu fréquente (1x/semaine – 1x/mois) pour certaines molécules, utilisation pour l’hémophilie A et B avec ou sans inhibiteur, potentielle utilisation pour d’autre(s) déficit(s) en facteurs de la coagulation. Les effets secondaires thrombotiques de ces deux approches doivent être pris en compte tels qu’en attestent le décès d’un patient traité par Fitusiran des suites d’une thrombose veineuse cérébrale et la survenue récente de thromboses artérielles chez des patients traités par anti-TFPI. Alors que les études sont en cours, il est difficile d’anticiper quel sera le succès et la place dans l’avenir de ces agents thérapeutiques dans la prise en charge future de l’hémophilie.
Les premiers succès prometteurs de la thérapie génique
Les progrès sont actuellement majeurs tant pour l’hémophilie A que pour l’hémophilie B. La stratégie utilisée consiste à insérer dans les hépatocytes le gène du FVIII ou du FIX. Elle fait appel à des vecteurs viraux à tropisme hépatique (adeno-associated virus ou AAV) (Figure 7). Les études en cours utilisent des sérotypes différents et des séquences génétiques optimisées pour favoriser l’expression et la synthèse du FVIII et du FIX (23).
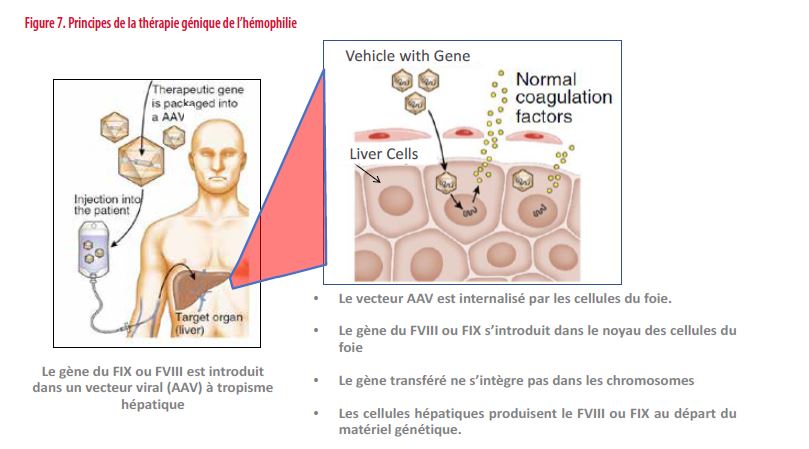
Sur base des quelques études dont les résultats sont publiés, la plupart des patients traités ne saignent quasi plus, ne doivent plus être traités par concentrés de facteurs et obtiennent des concentrations de FVIII et FIX variables d’un patient à l’autre, parfois normalisées (> 50 %), mais le plus souvent comprises entre 20-40 %. Ce résultat semble se maintenir dans le temps pour l’HB. Par contre, après 3 ans de recul, il semble y avoir une perte progressive d’expression dans le temps pour l’HA chez plusieurs patients (24;25).
Même si la thérapie génique semble prometteuse, plusieurs obstacles demeurent. Elle n’est actuellement pas accessible aux enfants. Certains patients ne peuvent être traités parce que porteurs d’anticorps neutralisants dirigés contre le vecteur viral. La survenue chez certains patients d’une réaction immunitaire dirigée contre des antigènes viraux exprimés à la surface des hépatocytes transfectés impose le recours à une corticothérapie transitoire. Nous ignorons actuellement combien de temps persistera l’expression des facteurs et surtout si le matériel génétique ne s’intégrera pas dans l’ADN des patients traités en y provoquant des erreurs génétiques.
Plusieurs centaines de patients sont actuellement inclus dans de multiples études dont les résultats devraient être communiqués en 2020 et nous éclairer à large échelle sur les succès et la tolérance de la thérapie génique.
L’enjeu de la participation aux études cliniques
Des progrès majeurs sont actuellement en cours de concrétisation dans le domaine du traitement de l’hémophilie. Il va de soi que ces traitements ne pourront être validés et voir le jour que si suffisamment de patients acceptent de contribuer aux études cliniques. Il existe plusieurs initiatives pour sensibiliser les patients à l’importance d’une participation active dans les études cliniques, en tenant compte de la contrainte et des risques éventuels qui doivent ouvertement discutés avec chaque patient (26).
Les incertitudes liées aux innovations thérapeutiques
Les concentrés de FVIII et FIX standards ou modifiés facilitent le traitement de l’hémophilie, surtout pour le FIX. Ils devraient largement remplacer les concentrés de FVIII et FIX « standards ». L’immunogénicité des concentrés de FVIII à demi-vie prolongée doit toutefois encore être bien explorée, surtout chez les jeunes enfants en début de traitement.
Les thérapies non substitutives s’avèrent efficaces pour prévenir les hémorragies et offrent l’avantage d’une administration sous-cutanée et peu fréquente. Toutefois, leur utilisation à l’occasion de gestes invasifs (chirurgie), leur monitoring biologique et les risques thrombotiques représentent autant de préoccupations (27).
Si la thérapie génique s’avère prometteuse, la réaction hépatique, la durabilité de l’effet et la sécurité au long court doivent être évaluées.
Sécurité et information objective : deux préoccupations majeures
Alors que les innovations thérapeutiques se multiplient, la sécurité des patients hémophiles est une priorité absolue. Chaque patient doit avoir accès à une information objective des réels bénéfices et des potentiels risques de tout nouveau traitement (28). La communauté hémophile doit de son côté faire preuve d’ouverture face aux diverses innovations et aux changements qu’elles entraînent.
Plus que jamais, chaque patient doit bien connaître et comprendre sa maladie et son traitement. Une réelle capacité d’apprentissage à gérer son traitement actuel et futur doit être promue. Entre patients, il est important de partager ses expériences thérapeutiques et de témoigner. Les patients hémophiles à travers les associations nationales et globales de patients doivent pouvoir exprimer leurs souhaits et leurs attentes pour que les innovations répondent à leurs aspirations et soient adoptées. Finalement, on peut émettre l’espoir que ces nouvelles options thérapeutiques prometteuses permettront d’améliorer la prise en charge des hémophiles dans le monde.
Que retenir ?
L’administration régulière et prophylactique des facteurs manquants de la coagulation (FVIII et FIX) représente le traitement actuel de référence de l’hémophilie. Cette approche est limitée par la contrainte d’injections intraveineuses fréquentes, son efficacité imparfaite et le développement d’anticorps neutralisants les FVIII et FIX. De nombreuses innovations devraient apporter des solutions. Il s’agit du développement de facteurs à demi-vie prolongée. S’y ajoutent divers agents administrés par voie sous-cutanée et moins fréquemment, tels qu’un anticorps bispécifique mimant l’action du FVIII et plusieurs molécules réduisant l’inhibition physiologique de la coagulation. Les premiers résultats de thérapie sont par ailleurs prometteurs. La bonne utilisation des nouveaux traitements disponibles et la validation des approches les plus récentes en garantissant la sécurité des patients représentent des défis importants.
Références
1. Peyvandi F, Garagiola I, Young G. The past and future of haemophilia: diagnosis, treatments, and its complications. Lancet 2016; 388(10040):187-197.
2. Oldenburg J. Optimal treatment strategies for hemophilia: achievements and limitations of current prophylactic regimens. Blood 2015; 125(13):2038-2044.
3. Butterfield JSS, Hege KM, Herzog RW, Kaczmarek R. A Molecular Revolution in the Treatment of Hemophilia. Mol Ther 2019.
4. Pelland-Marcotte MC, Carcao MD. Hemophilia in a Changing Treatment Landscape. Hematol Oncol Clin North Am 2019; 33(3):409-423.
5. Franchini M, Mannucci PM. Past, present and future of hemophilia: a narrative review. Orphanet J Rare Dis 2012; 7:24.
6. Kumar R, Dunn A, Carcao M. Changing Paradigm of Hemophilia Management: Extended Half-Life Factor Concentrates and Gene Therapy. Semin Thromb Hemost 2016; 42(1):18-29.
7. Franchini M, Mannucci PM. Non-factor replacement therapy for haemophilia: a current update. Blood Transfus 2018; 16(5):457-461.
8. Weyand AC, Pipe SW. New therapies for hemophilia. Blood 2019; 133(5):389-398.
9. Sankar AD, Weyand AC, Pipe SW. The evolution of recombinant factor replacement for hemophilia. Transfus Apher Sci 2019.
10. Moreno MM, Cuesta-Barriuso R. A history of prophylaxis in haemophilia. Blood Coagul Fibrinolysis 2019; 30(1S Suppl 1):S1-S3.
11. Dargaud Y, Delavenne X, Hart DP, Meunier S, Mismetti P. Individualized PK-based prophylaxis in severe haemophilia. Haemophilia 2018; 24 Suppl 2:3-17.
12. Pasca S, Zanon E. Savings without changing: How to use the MyPKfit(R) device to improve treatment strategies in a cohort of patients with haemophilia A. Thromb Res 2019; 183:1-3.
13. Iorio A, Edginton AN, Blanchette V, Blatny J, Boban A, Cnossen M et al. Performing and interpreting individual pharmacokinetic profiles in patients with Hemophilia A or B: Rationale and general considerations. Res Pract Thromb Haemost 2018; 2(3):535-548.
14. Makris M. Hemophilia A treatment: disruptive technology ahead. Blood 2016; 127(13):1623-1624.
15. Mahlangu J. Emicizumab for the prevention of bleeds in hemophilia A. Expert Opin Biol Ther 2019; 19(8):753-761.
16. Blair HA. Emicizumab: A Review in Haemophilia A. Drugs 2019.
17. Franchini M, Marano G, Pati I, Candura F, Profili S, Veropalumbo E et al. Emicizumab for the treatment of haemophilia A: a narrative review. Blood Transfus 2019; 17(3):223-228.
18. Susen S, Gruel Y, Godier A, Harroche A, Chambost H, Lasne D et al. Management of bleeding and invasive procedures in haemophilia A patients with inhibitor treated with emicizumab (Hemlibra((R)) ): Proposals from the French network on inherited bleeding disorders (MHEMO), the French Reference Centre on Haemophilia, in collaboration with the French Working Group on Perioperative Haemostasis (GIHP). Haemophilia 2019; 25(5):731-737.
19. Makris M, Iorio A, Lenting PJ. Emicizumab and thrombosis: The story so far. J Thromb Haemost 2019; 17(8):1269-1272.
20. Pasi KJ, Rangarajan S, Georgiev P, Mant T, Creagh MD, Lissitchkov T et al. Targeting of Antithrombin in Hemophilia A or B with RNAi Therapy. N Engl J Med 2017; 377(9):819-828.
21. Chowdary P. Anti-tissue factor pathway inhibitor (TFPI) therapy: a novel approach to the treatment of haemophilia. Int J Hematol 2018.
22. Shapiro AD, Angchaisuksiri P, Astermark J, Benson G, Castaman G, Chowdary P et al. Subcutaneous concizumab prophylaxis in hemophilia A and hemophilia A/B with inhibitors: Phase 2 trial results. Blood 2019.
23. Gollomp KL, Doshi BS, Arruda VR. Gene therapy for hemophilia: Progress to date and challenges moving forward. Transfus Apher Sci 2019.
24. Peyvandi F, Garagiola I. Clinical advances in gene therapy updates on clinical trials of gene therapy in haemophilia. Haemophilia 2019.
25. Pasi KJ, Rangarajan S, Mitchell N, Lester W, Symington E, Madan B et al. Multiyear Follow-up of AAV5-hFVIII-SQ Gene Therapy for Hemophilia A. N Engl J Med 2020; 382(1):29-40.
26. Henrard S, Speybroeck N, Hermans C. Participation of people with haemophilia in clinical trials of new treatments: an investigation of patients’ motivations and existing barriers. Blood Transfus 2015; 13(2):302-309.
27. Makris M, Hermans C. A golden age for Haemophilia treatment? Haemophilia 2018; 24(2):175-176.
28. Miesbach W, O’Mahony B, Key NS, Makris M. How to discuss gene therapy for haemophilia? A patient and physician perspective. Haemophilia 2019; 25(4):545-557.
Correspondance
Pr. Cedric Hermans, MD, PhD, FRCP (Lon, Edin)
Cliniques universitaires Saint-Luc
Service d’hématologie
Avenue Hippocrate 10
B-1200 Brussels BELGIUM
cedric.hermans@uclouvain.be