L’adénome hypophysaire, nouvellement appelé tumeur neuroendocrine pituitaire (PiNET) d’après la nouvelle classification oncologique de l’Organisation mondiale de la santé (OMS) parue en 2017 (1), fait partie des deux tumeurs intracrâniennes les plus fréquemment diagnostiquées : il compte effectivement pour 15% de celles-ci (2). On estime, sur base de 21604 autopsies réalisées, que 10% de la population est porteuse d’un adénome hypophysaire (3): la quasi-totalité (99.9%) des tumeurs décrites dans cette série est constituée de microadénomes dont la taille, par définition, est inférieure à 10 mm. Ces microadénomes s’opposent aux macroadénomes (dont la taille minimale est de 10mm) et représentent 52% de l’ensemble de ces tumeurs. Une personne sur 1100 se voit diagnostiquer, au cours de son existence, un adénome hypophysaire (4).
Endocrinologie
La première étape dans la mise au point diagnostique est hormonale. Une simple prise de sang permet de définir deux types d’adénomes : les tumeurs cliniquement fonctionnelles qui sécrètent une hormone en excès et les tumeurs non fonctionnelles qui ne sécrètent pas d’hormone de façon pathologique. Les adénomes non fonctionnels représentent 30% des adénomes hypophysaires diagnostiqués. Ils se manifestent essentiellement en raison de son effet de masse (4): il peut provoquer un déficit du champ visuel (18-78% des patients, classiquement une hémianopsie bitemporale par compression de la portion centrale du chiasma optique surplombant la loge pituitaire), des céphalées (17-75% des patients) ou une insuffisance hormonale anté-hypophysaire plus ou moins complète (34-89% des cas) (4).
Un screening biologique hormonal complet est donc essentiel : les tumeurs neuroendocrines pituitaires fonctionnelles engendrent un tableau clinique en rapport avec le type d’hormone produite en excès ; par ordre décroissant de fréquence, on retrouve (Tableau 1) :
1. les adénomes lactotropes ou prolactinomes: ils représentent 53% de tous les adénomes hypophysaires avec une prévalence d’environ 54 pour 100000 personnes et sont 10 fois plus fréquents chez la femme que chez l’homme. Il s’agit en majorité de microadénomes, les macro-prolactinomes étant plus fréquents dans la population masculine (50 à 75% contre 9 à 27% chez les femmes) (6,7). Environ 93% des patientes porteuses développent une oligoménorrhée et 50-85% d’entre elles, une galactorrhée (6,7) ;
2. les adénomes somatotropes (GH): ils provoquent une acromégalie clinique, essentiellement secondaire aux effets de l’insulin growth factor 1 (IGF-1) produite par le foie en réponse à la GH. Ils comptent pour 12% des adénomes, soit une prévalence de 10 cas par million d’habitants. Il s’agit en grande majorité (70%) de macroadénomes, souvent invasifs au moment du diagnostic et une co-sécrétion de prolactine n’est pas rare (30-40%) (7) ;
3. les adénomes corticotropes (ACTH): ils sont responsables de la maladie de Cushing caractérisée par un hypercorticisme causant un faciès pléthorique dit « en lune » avec fragilité cutanée et stries violacées, une obésité centrale avec hypertension artérielle, intolérance glucidique ou diabète, ostéoporose et perturbation du cycle menstruel féminin. Ces tumeurs constituent environ 4% des adénomes hypophysaires, soit 1.6 cas par million de personnes. On note une prédominance féminine (8 cas pour 1 homme) et prennent majoritairement (90%) la forme de microadénome (7,8) ;
4. les adénomes thyréotropes (TSH): 1% des adénomes, 75% de macro-adénomes, provoquant une hyperthyroïdie (75%), un goitre (55%), une fibrillation auriculaire ou décompensation cardiaque (11%) et sécrétant en parallèle la PRL ou la GH (32% (9,10).
La plupart du temps, un bilan biologique hormonal simple permet d'établir le diagnostic. Le diagnostic du syndrome de Cushing nécessite parfois des mesures répétées hormonales sur le nycthémère et des tests dynamiques. Dans d’autres cas, lorsque ces valeurs sériques sont « borderline », certains tests spécifiques sont nécessaires et nécessitent une courte hospitalisation : c’est le cas du test de tolérance au glucose, indispensable au diagnostic d’une acromégalie, lorsque le taux sérique d’IGF-1 n’est que de 1.1 à 1.5 fois supérieure à la limite normale supérieure. Le manque de suppression de la GH (<0.4µg/L) après ingestion d’une quantité orale de 75g de glucose est sensible à 85-90% et spécifique à >95% (11-13). Cette hospitalisation est mise à profit pour détecter les éventuelles complications systémiques de l’acromégalie : arthropathie (70%), syndrome d’apnée du sommeil (65%), syndrome du canal carpien (64%), diabète ou prédiabète (50% des patients), hypertension artérielle (35%), valvulopathie cardiaque (30%) et cardiomyopathie (10%), polypes coliques (32%). Certains adénomes augmentent le risque relatif plus important de cancer, notamment colique, mais également thyroïdien, mammaire ou urologique (7,14,15)
Imagerie médicale
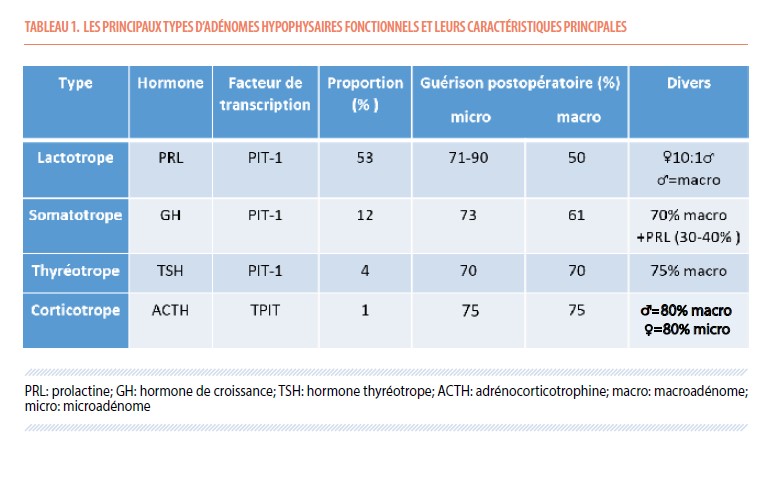
Après cette confirmation endocrinologique, une mise au point par résonance magnétique (IRM) de qualité (3-Tesla) centrée sur la loge hypophysaire est primordiale : des séquences spécifiques seront réalisées, essentiellement des coupes coronales fines avant et après injection de gadolinium par voie intraveineuse, ainsi qu’une séquence T2 et des séquences dites « dynamiques ». Une imagerie de qualité permettra de localiser précisément les microadénomes au sein de la glande pituitaire et d’analyser l’extension extra-sellaire des macro-adénomes (16). En particulier, l’invasion du sinus caverneux sera déterminante dans la qualité de résection chirurgicale de ces adénomes supra-centimétriques : cette loge veineuse constitue, de chaque côté, la limite latérale de la loge hypophysaire et contient l’artère carotide homolatérale ainsi que les 3 nerfs oculomoteurs (III, IV, VI) et 2 branches du nerf trijumeau (V1, V2). La probabilité d’invasion de ce sinus est évaluable sur les coupes coronales de l’IRM selon la classification de Knosp (17) (Figure 1): plus le grade est élevé, plus l’invasion caverneuse est probable, plus le risque de résidu tumoral postopératoire est grand, moins la probabilité de guérison est importante.
Ophtalmologie
La dernière étape de la mise au point est la consultation ophtalmologique : une évaluation précise du champ visuel afin de mettre en évidence une éventuelle compression du chiasma optique est primordiale. En cas d’atteinte visuelle, un traitement chirurgical rapide sera vivement recommandé au patient.
Concertation multidisciplinaire oncologique (CMO)
Chaque semaine, les dossiers des patients porteurs d’une tumeur neuroendocrine pituitaire sont discutés en réunion multidisciplinaire dite « oncologique » : les radiologues, les endocrinologues et les neurochirurgiens se mettent d’accord, sur base des dernières guidelines, du traitement le plus approprié à proposer au patient (18). De manière générale, les macroadénomes non fonctionnels compressifs sur les voies visuelles ou de taille importante, les adénomes à GH, à ACTH, à TSH seront opérés, tandis que les prolactinomes (micro ou macro) seront d’abord traités de façon médicamenteuse. Effectivement, les analogues dopaminergiques (cabergoline en première intention) ont la capacité potentielle de normaliser les taux sériques de prolactine, de traiter leurs symptômes évoqués précédemment et même de réduire drastiquement leur volume tumoral (jusqu’à 65% des cas) (7,19). Seuls les patients résistants à ce traitement médicamenteux et ceux en subissant trop d’effets secondaires (2.5 à 50% des patients rapportent ces effets indésirables pour la cabergoline, les plus fréquents étant nausées, hypotension orthostatique, céphalées, dépression et vasospasme digital) (20,21) se verront proposer une intervention chirurgicale. Finalement, les patients porteurs d’adénomes non fonctionnels de petite dimension se verront proposer une surveillance clinique et radiologique. Parmi ceux-ci, on retrouve les incidentalomes pituitaires : ces lésions adénomateuses hypophysaires découvertes fortuitement suite à une IRM cérébrale réalisée pour un autre motif sont de plus en plus fréquentes. Parmi celles-ci, approximativement 10% des microlésions et 25% des macrolésions grandissent au cours du temps. En cas de croissance significative, une résection chirurgicale pourra être proposée au patient (22).
Neurochirurgie
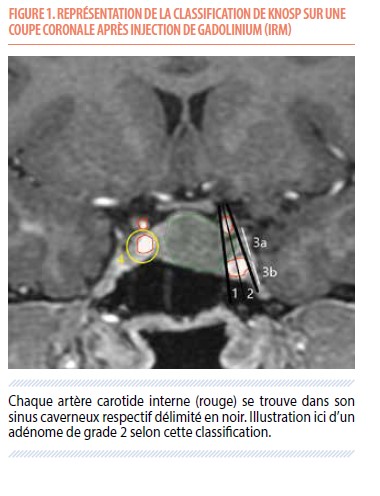
Le patient entrera à l’hôpital la veille de l’intervention chirurgicale et une nouvelle IRM sera réalisée afin de planifier au mieux la résection tumorale. Cette chirurgie se fera sous anesthésie générale, par voie transnasale, sous microscopie opératoire et à l’aide de la neuro-navigation, un véritable système GPS aidant le chirurgien à obtenir l’exérèse la plus complète possible. Un modèle de planification en 3 dimensions est représenté sur la Figure 2. Le temps opératoire est inférieur à une heure. Il restera ensuite 4 jours complémentaires dans l’unique but d’une surveillance endocrinologique : la complication hormonale postopératoire la plus fréquente est la survenue d’un diabète insipide (polyurie-polydipsie, 5%) : un traitement substitutif par desmopressine sera alors instauré. La plupart du temps, le diabète insipide est transitoire et résolution complète est constatée dans 90% des cas au cours des semaines suivantes (4). Un contrôle méticuleux de la diurèse sera donc réalisé, tout comme un ionogramme (prise de sang) au 3e jour postopératoire, puis au 7e jour postopératoire, le patient étant déjà sorti de l’hôpital. Au 4e jour postopératoire, un contrôle biologique sanguin de tous les axes hormonaux est également effectué afin de vérifier l’efficacité chirurgicale pour les adénomes fonctionnels, mais aussi la survenue d’un éventuel nouveau déficit hormonal (5 à 15% des patients) (4).
Suivi
Une consultation de contrôle en endocrinologie est prévue 6 semaines après l’intervention ; un nouveau screening sanguin hormonal complet y est réalisé. Un suivi par IRM est ensuite effectué à 3 mois de l’opération afin d’exclure un résidu tumoral (23). Le patient rencontre alors à nouveau le neurochirurgien et son dossier est rediscuté en CMO. Certaines données récentes remettent en question ce délai de 3 mois et optent pour une IRM de contrôle endéans les 3 jours suivant la chirurgie : ce délai serait plus sensible pour détecter une masse adénomateuse résiduelle (24). Les grandes séries de la littérature estiment à 65% le taux de résection complète des macro-adénomes non fonctionnels avec une résolution des fonctions visuelles pouvant aller jusqu’à 90% des cas (4). La résolution des déficits hormonaux préopératoires peut quant à elle survenir dans 50% des cas (25,26). Les taux de guérison après chirurgie des adénomes fonctionnels sont estimés comme suit (Tableau 1) : 71-90% pour les microprolactinomes, 50% pour les macroprolactinomes (27), 73% pour les microadénomes à GH, 61% pour les macroadénomes à GH (28, 29, 30), environ 70% pour les adénomes à TSH (9,10) et 75% pour les adénomes à ACTH (31). Concernant ces derniers, notre taux de guérison est de 70% après une première intervention et de 83% après une ou plusieurs chirurgies (32).
Le suivi à moyen et long terme se fait essentiellement de manière biologique et clinique pour les adénomes fonctionnels et de façon radiologique pour les adénomes non fonctionnels. En cas de récidive ou de croissance du résidu tumoral, une nouvelle chirurgie ou une radiothérapie ciblée pourra être proposée au patient après rediscussion multidisciplinaire. Un diagramme résumé de la prise en charge globale des adénomes hypophysaires au sein des Cliniques Universitaires Saint-Luc est représenté sur la Figure 3.

Cas clinique
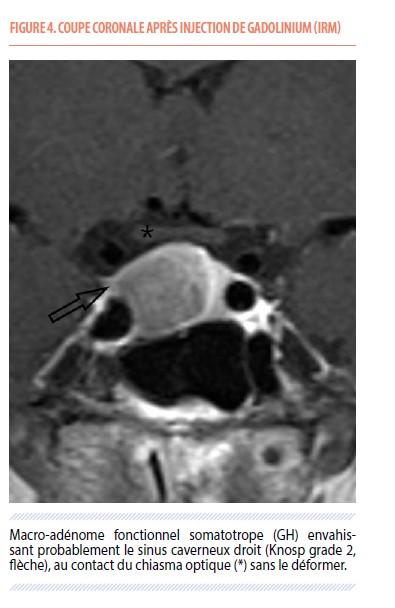
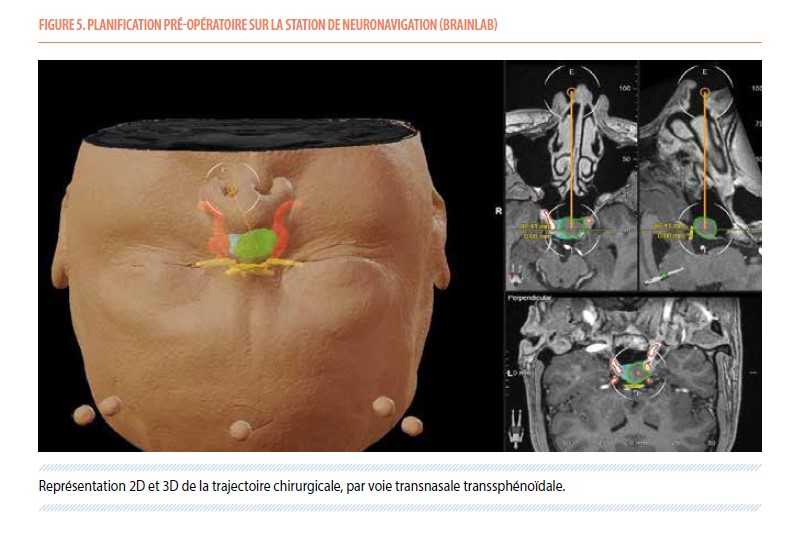
Madame E.C., sans antécédents médicaux notables, se présente à la consultation d’endocrinologie après la découverte d’un probable macroadénome hypophysaire, à la suite d’une IRM cérébrale réalisée pour un motif de céphalées. Le premier bilan biologique hormonal montre un taux sérique d’IFG-1 à 737.9 ng/mL (valeurs normales entre 131 et 320 ng/mL). Le reste du bilan hormonal est normal hormis une légère insuffisance thyréotrope (TSH normale, T4 libre à 10 pmol/L pour des valeurs normales entre 12 et 22) pour laquelle un traitement oral par hormones thyroïdiennes est instauré. À l’anamnèse, on retrouve une croissance progressive de la taille de ses mains, des cycles menstruels devenus irréguliers et une prise pondérale de 20kg au cours des 5 années précédentes. Elle présente également des paresthésies, essentiellement nocturnes, au niveau des deux mains et des œdèmes aux membres inférieurs confirmés à l’examen clinique. On note un indice de masse corporelle (IMC) à 29 kg/m². Une courte hospitalisation permet de détecter de potentielles complications de l’acromégalie : colonoscopie normale, oxymétrie nocturne en défaveur d’un syndrome d’apnée du sommeil, discrète insuffisance mitrale. Un profil nycthéméral de la cortisolémie et de l’ACTH met en évidence une discrète insuffisance corticotrope. Le champ visuel est normal. Une IRM dédiée à l’hypophyse retrouve un macroadénome hypophysaire de 17x15x14mm en contact avec le chiasma optique qui n’est cependant pas déformé, une déviation de la tige pituitaire vers la gauche et une possible invasion du sinus caverneux droit (Knosp de grade 2) (Figure 4). Le dossier de madame E.C. est alors discuté en CMO qui conclut à une résection chirurgicale sans urgence. Les tenants et aboutissants potentiels de cette intervention lui sont ensuite expliqués en consultation de neurochirurgie et la patiente accepte l’opération. Une voie transnasale transsphénoïdale sous microscope opératoire et neuronavigation (Figure 5) permet alors une résection évaluée environ 90% de la masse tumorale, sans complications. Six semaines plus tard, en consultation d’endocrinologie, on constate une normalisation du taux sérique de l’IGF-1 (266 ng/mL) signant une rémission de l’acromégalie. L’examen d’anatomopathologie confirme un adénome hypophysaire à GH avec un index mitotique (taux de division cellulaire) de 5%.
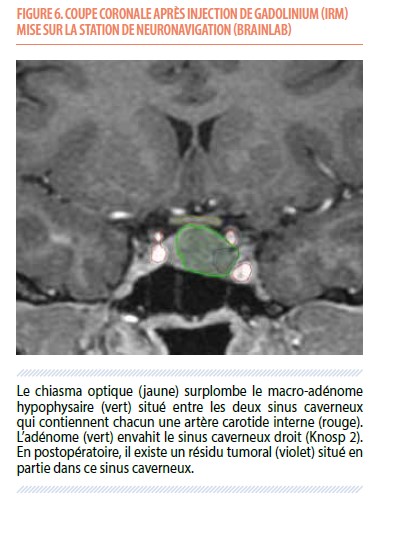
L’IRM postopératoire à 3 mois révèle un résidu tumoral mesurant environ 10% du volume tumoral initial : il est situé au niveau du sinus caverneux droit, confirmant l’invasion initiale de celui-ci (Figure 6). Son suivi hormonal sera marqué par une récidive a minima de l’acromégalie biologique environ 13 mois après la chirurgie (351.7 ng/mL); récidive confirmée par un test dynamique d’ingestion de glucose montrant une suppression insuffisante de la GH. Après une nouvelle discussion multidisciplinaire, on instaure un traitement médicamenteux par cabergoline: cet analogue dopaminergique, traitement préférentiel des prolactinomes, est parfois efficace pour traiter les adénomes à GH (33). Ce traitement a effectivement permis de normaliser les taux sériques d’IGF-1 durant 4 années complémentaires, ainsi qu’une stabilité du résidu tumoral (Figure 7).

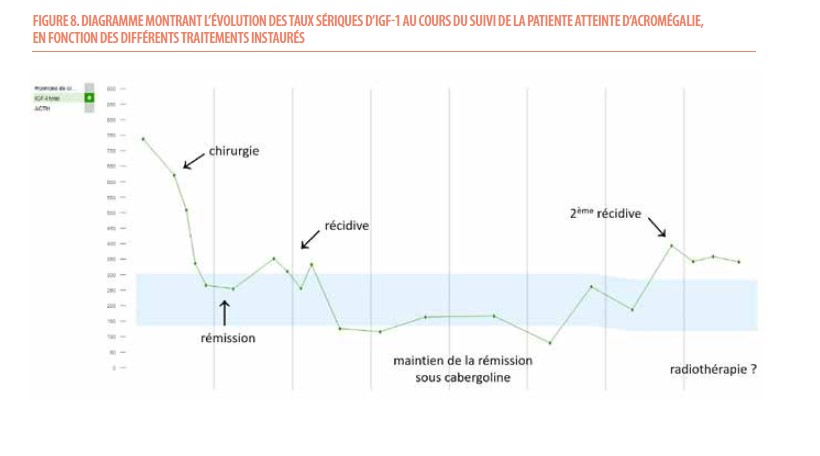
Actuellement, il existe une nouvelle récidive biologique a minima (393.6 ng/mL); le résidu est toujours stable à l’IRM. Une radiothérapie sur ce morceau tumoral persistant sera prochainement discutée en CMO. Le taux sérique d’IGF-1 de cette patiente au cours du temps est montré sur la Figure 8.
Conclusion
Les adénomes hypophysaires ou tumeurs neuroendocrines pituitaires constituent l’une des deux tumeurs bénignes intracrâniennes les plus fréquentes. Ils peuvent provoquer un syndrome endocrinologique d’hypersécrétion hormonale, un déficit visuel ou une insuffisance hypophysaire. Une mise au point pluridisciplinaire (endocrinologie, radiologie et ophtalmologie) est primordiale et leur traitement est essentiellement neurochirurgical. Au sein d’une équipe entraînée, les résultats sont très bons, tant en termes de contrôle hormonal qu’en termes de levée de l’effet de masse. Les microprolactinomes sont les plus fréquents parmi ces tumeurs et se traitent en première intention par la prise orale d’analogues dopaminergiques tels que la cabergoline. Un suivi biologique et radiologique est essentiel tout au long de la vie des patients porteurs d’un adénome hypophysaire.
Affiliations
1. Service de Neurochirugie des Cliniques Universitaires Saint-Luc
2. Service d’Endocrinologie des Cliniques Universitaires Saint-Luc
Correspondance
Pr Edward FOMEKONG
Cliniques universitaires Saint-Luc
Service de Neurochirurgie
Avenue Hippocrate 10
B-1200 Bruxelles
edward.fomekong@saintluc.uclouvain.be
Références
- Asa S.L, Casar-Borota O, Karavitaki N., et al. From pituitary adenoma to pituitary neuroendocrine tumor (PitNET): An International Pituitary Pathology Club proposal. Endocr. Relat. Cancer. 2017 ; 24:C5–C8. doi: 10.1530/ERC-17-0004
- Gittleman H, Ostrom QT, Farah PD et al. Descriptive epidemiology of pituitary tumor in the United States, 2004-2009. J Neurosurg. 121:527-535, 2014. doi: 10.3171/2014.5.JNS131819
- Molitch ME. Nunfunctioning pituitary tumors. Handb Clin Neurol. 2014:124:167-184. doi: 10.1016/B978-0-444-59602-4.00012-5
- Tritos NA, Miller KK. Diagnosis and management of pituitary adenoma: a review. JAMA. 2023 ;329(16):1386-1398. doi:10.1001/jama.2023.5444
- Molitch ME. Nunfunctioning pituitary tumors and pituitary incidentalomas. Endocrinol Metab Clin North Am. 2008;37(1):151-171. doi: 10.1016/j.ecl.2007.10.011
- Klibanski A. Clinical practice: prolactinomas. N Engl J Med. 2010;362(13):1219-1226. doi:10.1056/ NEJMcp0912025
- Shlomo M. Pituitary-tumors endocrinopathies. N Engl J Med. 2020;382:937-50. doi: 10.1056/NEJMra1810772
- Lacroix A, Feelders RA, Stratakis CA et al. Cushing’s syndrome. Lancet. 2015;386(9996):913-917. doi: 10.1016/ S0140-6736(14)61375-1
- Amlashi FG, Tritis NA. Thyrotropin-secreting pituitary adenomas: epidemiology, diagnosis, and management. Endocrine. 2016;52(3):427-440. doi: 10.1007/s12020-016-0863-3
- De Herdt C, Philipse E, De BlockC. Endocrine tumors: thyrotropin-secreting pituitary adenoma: a structured reviex of 553 adult cases. Eur J Endocrinol. 2021;185(2):R65-R74. doi: 10.1530/EJE-21-0162
- Katznelson L, Laws ER, Melmed S et al. Endocrine Society. Acromegaly: an endocrine society clinical practice guidelines. J Clin Endocrinol Metab. 2014;99(11):3933:3952. doi: 10.1210/jc.2014-2700
- Ribeiro-Oliveira A Jr, Faje AT, Brakan AL. Limited utility of oral glucose tolerance test in biochemically active acromegaly. Eur J Endocrinol. 2011;164(1):17-22. doi: 10.1530/EJE-10-0744
- Dobri G, Niwattisaiwong S, Bena JF et al. Is GH nadir during OGTT a reliable test for diagnosis of acromegaly in patients with abnormal glucose matabolism? Endocrine. 2019;64(1):139-146. doi: 10.1007/s12020-018-1805-z
- Ben-Shlomo A, Melmed S. Acromegaly. Endocrinol Metab Clin North Am. 2008;37(1):101-122. doi: 10.1016/j.ecl.2007.10.002
- Dal J, Leisner MZ, Hermansen K, et al. Cancer incidence in patients with acromegaly: a cohort study and meta-analysis of the literature. J Clin Endocrinol Metab. 2018;103: 2182-8. doi: 10.1210/jc.2017-02457
- MacFarlane J, Bashari WA, Senanayake R et al. Advances in the imaging of pituitary tumors. Endocrinol Metab Clin North Am. 2020;49(3):357-373. doi: 10.17863/CAM.55828
- Araujo-Castro M, Acitores Cancela A, Rodriguez Berrocal V et al. Radiological Knosp, revides-Knosp and Hardy-Wilson classifications for the prediction of surgical outcomes in the endoscopic endonasal surgery of pituitary adenomas: study of 228 cases. Front Oncol. 2022. 11:807040. doi: 10.3389/fonc.2021.807040
- Domingue ME, Alexopoulo O, Maier D. Prise en charge multidisciplinaire des adénomes hypophysaires en 2013. Louvain Med. 2013;132(2):64-7
- Biller BM, Molitch ME, Vance ML, et al. Treatment of prolactin-secreting macro adenomas with the once-weekly dopamine agonist cabergoline. J Clin Endocrinol Metab. 1996; 81: 2338-43. doi: 10.1210/jcem.81.6.8964874
- Wang AT, Mullan RJ, Lane MA et al. Treatment of hyperprolactinemia: a systematic review and meta-analysis. Syst Rev. 2012;1(1):33. doi: 10.1186/2046-4053-1-33
- Ioachimescu AG, Fleseriu M, Hoffman AR, Vaughan Iii TB, Katznelson L. Psychological effects of dopamine agonist treatment in patients with hyperprolactinemia and prolactin-secreting adenomas. Eur J Endocrinol. 2019; 180: 31-40. doi: 10.1530/EJE-18-0682
- Constantinescu SM, Maiter D. Pituitary incidentaloma. Presse Med. 2021; 50(4):104081. doi: 10.1016/j.lpm.2021.104081
- Ziu M, Dunn IF, Hess C, Fleseriu M, Bodach ME, Tumialan LM, et al: Congress of Neurological surgeons systematic review and evidence-based guideline on posttreatment follow-up evaluation of patients with nonfunctioning pituitary adenomas. Neurosurgery. 2016:79:E541–E543. doi: 10.1227/NEU.0000000000001392
- Alhilali LM, Little AS, White WL et al. Early postoperative MRI and detection of residual adenoma after transsphenoidal pituitary surgery. J Neurosurg. 134:761–770, 2021. doi : 10.3171/2019.11.JNS191845
- Chen L, White WL, Spetzler RF, Xu B.A prospective study of nonfunctioningpituitary adenomas: presentation, management,and clinical outcome. J Neurooncol. 2011; 102: 129-38. doi: 10.1007/s11060-010-0302-x
- Paluzzi A, Fernandez-Miranda JC, Tonya Stefko S, Challinor S, Snyderman CH, Gardner PA. Endoscopic endonasal approach for pituitary adenomas: a series of 555 patients. Pituitary. 2014; 17: 307-19. doi: 10.1007/s11102-013-0502-4
- Tampourlou M, Trifanescu R, Paluzzi A, Ahmed SK, Karavitaki N. Therapy of endocrine disease: surgery in microprolactinomas: effectiveness and risks based on contemporary literature. Eur J Endocrinol. 2016; 175: R89-R96. doi: 10.1530/EJE-16-0087
- Buchfelder M, Schlaffer SM. The surgical treatment of acromegaly. Pituitary. 2017; 20: 76-83. doi: 10.1007/s11102-016-0765-7
- Kim JH, Hur KY, Lee JH, et al. Outcome of endoscopic transsphenoidal surgery for acromegaly. World Neurosurg. 2017; 104: 272-8. doi: 10.1016/j.wneu.2017.04.141
- Babu H, Ortega A, Nuno M, et al. Long-term endocrine outcomes following endoscopic endonasal transsphenoidal surgery for acromegaly and associated prognostic factors. Neurosurgery. 2017; 81: 357-66. doi: 10.1093/neuros/nyx020
- Nieman LK, Biller BM, Findling JW, et al. Treatment of Cushing’s syndrome: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab. 2015; 100: 2807-31. doi: 10.1210/jc.2015-1818
- Brichard C, Fomekong E, Raftopoulos C et al. Outcome of transsphenoidal surgery for Cushing disease: a single-center experience over 20 years. World Neurosurg. 2018 Nov;119:e106-e117. doi: 10.1016/j.wneu.2018.07.055
- Kuhn E, Chanson P. Cabergoline in acromegaly. Pituitary. 2017; 20: 121-8. doi: 10.1007/s11102-016-0782-6